
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Crystallization kinetics of thin water films on Pt(111): effects of oxygen and carbon-monoxide adspecies
Ryutaro
Souda
 a and
Takashi
Aizawa
b
a and
Takashi
Aizawa
b
aTransmission Electron Microscopy Station, National Institute for Materials Science, 1-2-1 Sengen, Tsukuba, Ibaraki 305-0047, Japan
bCenter for Functional Sensor & Actuator, National Institute for Materials Science, 1-1 Namiki, Tsukuba, Ibaraki 305-0044, Japan
First published on 4th January 2019
Abstract
This paper describes nucleation, epitaxial growth, and wettability of water on Pt(111) and how they are influenced by oxygen and carbon-monoxide adspecies, based on reflection high energy electron diffraction (RHEED), time-of-flight secondary ion mass spectrometry (TOF-SIMS), and temperature-programmed desorption (TPD). Amorphous solid water deposited onto the pristine Pt(111) substrate crystallizes into ice Ih together with a 2D layer at 150 K, whereas ice Ic (stacking disordered ice or a mixture of ice Ic and Ih) is formed preferentially onto oxygenated Pt(111) (CO-adsorbed Pt(111)) at 155–160 K (150 K). The ice nucleation and epitaxial growth tend to be hampered on the oxygenated Pt(111) surface via hydrogen bond formation with chemisorbed oxygen. The CO-adsorbed Pt(111) surface is hydrophobic, as evidenced by the fact that water forms a complex with CO during evaporation of crystallites at 160–165 K. A disordered 2D layer remains on pristine Pt(111) up to 175 K, whereas an ordered 2D layer exhibiting the (√3 × √3)R30° structure formed on oxygenated Pt(111) up to 200 K.
1 Introduction
Interfacial water is ubiquitous in nature and plays an important role in fundamental research and technological applications. Interactions of water with solid surfaces have been studied extensively at cryogenic temperatures in terms of wettability, chemical reaction, crystallization kinetics, etc.1,2 A considerable attention has been focused on water monolayers formed on close packed transition metal substrates because their lateral lattice parameter matches well with that of crystalline ices. A commensurate hexagonal ice bilayer, analogous to the (0001) plane of hexagonal ice Ih, is expected to form an ordered (√3 × √3)R30° structure, as was observed in low energy electron diffraction (LEED).1,2 However, the structure of thin crystalline ice films on a Pt(111) substrate is still controversial. In contrast to the conventional picture, it was depicted based on X-ray absorption measurements that all water molecules in the first layer bind directly to the Pt(111) surface via alternating metal–oxygen and metal–hydrogen bonds, thereby forming a flat H-down water monolayer.3 It was also revealed using helium atom scattering (HAS)4 and LEED5 that ordered 2D domains exhibiting (√37 × √37)R25° and (√39 × √39)R16° structures formed via a slow deposition of water molecules at a temperature of ca. 130 K; water forms hexagonal structures connected by pentagonal and heptagonal units, as imaged using scanning tunneling microscopy (STM).6 Although the water molecule does not dissociate on Pt(111),1,2,7 or does so in very small amounts,8 the (√3 × √3)R30° LEED pattern might be induced by chemisorbed O and OH species formed via electron-beam damage.9 In fact, a partially dissociated, hydrogen-bonded network of water exhibiting the (√3 × √3)R30° structure is known to be formed on a chemically more reactive substrate like Ru(0001).10 Consequently, it is likely that water forms a range of different hydrogen bonded clusters or networks on the Pt(111) surface,7 depending on the surface chemical reactivity that can be modified by a small amount of adspecies rather than the simple lattice parameter matching. At higher water coverage, continuous ice films can be formed on Pt(111): LEED I–V analyses revealed that crystalline films thicker than ca. 40–50 layers exhibited a (1 × 1) termination of ice Ih(0001).11 To date, crystallization kinetics has also been explored using thin films of amorphous solid water (ASW) based on many other experimental techniques, such as isothermal desorption,12–15 temperature programmed desorption (TPD),15–20 reflection high energy electron diffraction (RHEED),21–24 and reflection absorption infrared spectroscopy (RAIRS),19,25,26 together with the glass–liquid transition of ASW27,28 and premelting of crystalline ices28,29 based on time-of-flight secondary ion mass spectrometry (TOF-SIMS) and RHEED. In contradiction to the conventional picture of the ice Ih growth, it was revealed that ice Ic was formed preferentially on both hydrophilic and hydrophobic substrates based on the RHEED studies.21–24 The discrepancy might be related to the fact that small 3D crystallites descended from thinner ASW films can be analyzed by RHEED, in contrast to LEED that sees only top surfaces of wide spreads of 2D water layers and flat crystal grains. In this respect, RHEED is a much more powerful technique than LEED for studying nucleation and growth processes of crystalline ices.In this paper, we investigate the interactions of water with the Pt(111) substrate and the influence of adspecies to gain insight into how the crystallization kinetics and wetting of thin films are controlled by the interfacial properties. To this end, crystallization and dewetting kinetics of ASW on pristine, oxygenated, and CO-adsorbed Pt(111) substrates are explored especially in the monolayer regime by using RHEED, TOF-SIMS, and TPD as a function of substrate temperature. The structure and quality of crystalline ices formed by heating the initial ASW films are compared with those formed via water vapor deposition at 135 K based on the RHEED image.
2 Experiment
Experiments were performed in an ultrahigh vacuum (UHV) chamber (base pressure of 2 × 10−10 Torr) equipped with facilities of TOF-SIMS, RHEED and TPD. For TOF-SIMS measurements, a primary beam of 2 keV He+ ions was generated in a differentially-pumped electron-impact-type ion source (Specs GmbH, IQE 12/38); the ion beam was chopped into pulses using electrostatic deflectors. A bias voltage (+500 V) was applied to the sample surface, and a removable mesh plate (grounded) was placed in front of the sample to extract low-energy secondary ions efficiently. Positive secondary ions were detected using a microchannel plate after passing through a field-free TOF tube. TOF spectra were recorded every 30 s using a multichannel scaler (Laboratory Equipment Inc., LN-6500R). The fluence of He+ was kept below 1 × 1012 ions cm−2 (sample current of ∼0.1 nA cm−2) to minimize degradation of thin films. To obtain RHEED patterns, a focused high-energy electron beam (30 keV) was generated in an electron gun (Eiko Co. Ltd, MB-1000). The diffraction pattern on a phosphor screen was recorded using a high-sensitivity CCD camera intermittently (duration of ∼0.5 s) only at a specific temperature and coverage of interest to reduce the sample damage. TPD spectra were obtained using a differentially-pumped quadrupole mass analyzer (Hiden Analytical Ltd, IDP 300S). Temperature-programmed measurements were performed at a ramping speed of 5 K min−1.A mirror-finished Pt(111) substrate was spot-welded to a sample holder made of Ta. The substrate was heated to ∼1500 K by electron bombardment from behind. Contaminants were removed via several Ar+-sputtering and annealing cycles. Cooling of the sample was achieved by means of a closed-cycle helium refrigerator. The substrate temperature was controlled using a cartridge heater by monitoring the temperature of the cold finger close to the sample position using Au(Fe)-chromel thermocouples. Liquid water was degassed before use via several freeze–pump–thaw cycles. The H2O vapor and research-grade O2 and CO gases were admitted into the UHV chamber through precision variable leak valves. Cleanliness of the Pt(111) substrate with and without adspecies was ascertained based on the TOF-SIMS spectra. Regarding the ASW film thickness, the H+ and H3O+ intensities in TOF-SIMS were saturated at exposure to 2–3 langmuirs (1 L = 1 × 10−6 Torr s) at 100 K,29 where the first monolayer is expected to be formed. However, the sticking probability of water is likely to depend on the surface chemical properties and substrate temperature. In this paper, therefore, the films were characterized by water exposure rather than the exact coverage.
3 Experimental results
Fig. 1 shows (a) temperature-programmed TOF-SIMS intensities and (b) a TPD spectrum obtained using the ASW film prepared by deposition of 2 L water onto the Pt(111) substrate at 100 K (under a H2O backfilling pressure of 1 × 10−8 Torr). Upon adsorption of water, not only H+ and H+(H2O)n ions, but also ion adducts in the form of Pt+(H2O)n were emitted.29 Stepwise decreases are observed in the H+ and H3O+ intensities at 150, 160, and 170 K. The TPD spectrum exhibits a double-peak structure, corresponding to desorption from 3D (163 K) and 2D (170 K) domains of water on Pt(111).30,31 All secondary ions decay at 170 K along with the occurrence of the 2D peak in TPD. The first steps of H+ and H3O+ intensities at 150 K are ascribable to film dewetting during water crystallization (see the RHEED patterns described later). The second steps at 160 K result from desorption of crystallites, as manifested by the 3D peak in TPD. In contrast to H+ and H3O+ ions, the Pt+(H2O)2 ion survives considerably after disappearance of crystallites, because it is emitted preferentially from monolayer domains of water in direct contact with the Pt(111) substrate (i.e., the 2D layer). Note that a characteristic shoulder in TPD, which is known as a sign of crystallization of the multilayer ASW films,16,18 is not recognized at around 150 K.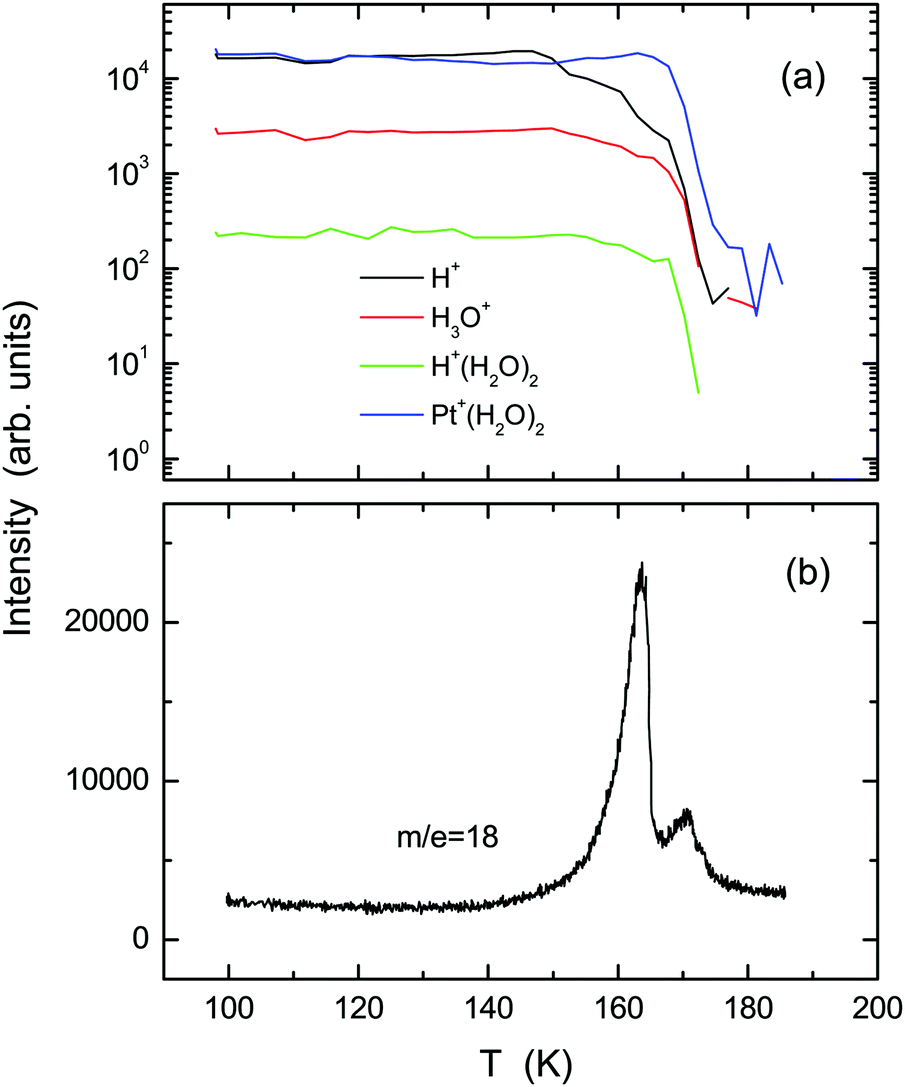 | ||
| Fig. 1 Temperature-programmed TOF-SIMS intensities (a) and the TPD spectrum (b) obtained using the ASW film formed on the Pt(111) substrate. The film was formed by exposure to 2 L H2O molecules at 100 K; then, the substrate temperature was increased at a rate of 5 K min−1. | ||
Upon heating above 150 K, the ASW film becomes inhomogeneous because of the growth of crystal grains. Their structure and epitaxial relationship with respect to the substrate are determined based on RHEED. Images taken along the [11![[2 with combining macron]](https://www.rsc.org/images/entities/char_0032_0304.gif) ] azimuth of the substrate are presented in Fig. 2. For the pristine Pt(111) surface (a), intense diffraction rods are recognizable in the zero-order and first-order Laue zones. After deposition of 2 L H2O molecules at 100 K (b), the rods from the Pt(111) substrate are weakened and diffuse scattering from ASW appears. The diffuse pattern is maintained fundamentally up to 140 K. Then, sharp streaks and Bragg spots characteristic of the crystalline ice are identifiable at temperatures of (c) 150, (d) 155, and (e) 160 K. No diffraction patterns other than those from the clean Pt(111) substrate are obtained at 165 K (f), indicating that the 2D layer formed immediately after evaporation of crystallites is not ordered.
] azimuth of the substrate are presented in Fig. 2. For the pristine Pt(111) surface (a), intense diffraction rods are recognizable in the zero-order and first-order Laue zones. After deposition of 2 L H2O molecules at 100 K (b), the rods from the Pt(111) substrate are weakened and diffuse scattering from ASW appears. The diffuse pattern is maintained fundamentally up to 140 K. Then, sharp streaks and Bragg spots characteristic of the crystalline ice are identifiable at temperatures of (c) 150, (d) 155, and (e) 160 K. No diffraction patterns other than those from the clean Pt(111) substrate are obtained at 165 K (f), indicating that the 2D layer formed immediately after evaporation of crystallites is not ordered.
 | ||
| Fig. 2 RHEED images of the pristine Pt(111) substrate (a) and the water-adsorbed Pt(111) substrate with increasing temperature (b–f). The ASW film was formed by deposition of 2 L water at 100 K; images obtained at temperatures of (b) 100, (c) 150, (d) 155, (e) 160, and (f) 165 K are displayed. They were taken along the [11−2] azimuth of Pt(111). | ||
It is expected that the (111) oriented ice Ic or the (0001) oriented ice Ih are grown on the Pt(111) substrate. Simulated RHEED patterns for the ices Ih and Ic, respectively, are shown in the left and right halves of Fig. 3(b), together with the indexed RHEED image in Fig. 3(a). The streaky RHEED patterns, which is close to the Pt(111) (√3 × √3)R30° periodicity, are basically ascribable to the ice crystallite. They consist of elongated Bragg spots because ice Ic and Ih structures coexist, specifically, the occurrence of stacking disorder in the basal planes.32 Additional sharp spots assigned as 114, 116, and 118 in Fig. 3(a) are attributable to crystallites grown in the other azimuthal direction, i.e., ±30° off relative to those yielding the (√3 × √3)R30°-like pattern. In this azimuth, the sharp spots of 3D crystallites are observed instead of the streaks because the Ic and Ih crystallites make spots at the same position. From these facts, the streaky patterns are better ascribable to the 3D grains of mixed ices Ic and Ih, or a stacking disordered ice, rather than a flat terrace of large ice Ih grains. This interpretation is compatible with the occurrence of dewetting (i.e., water aggregation to form 3D grains) during crystallization of the ASW film (Fig. 1(a)). The streaks underlying the Bragg spots suggest that crystal grains have a flat terrace or an ordered 2D water lattice coexists. The latter is discarded from the fact that streaks disappear after evaporation of crystallites, as seen in Fig. 2(f).
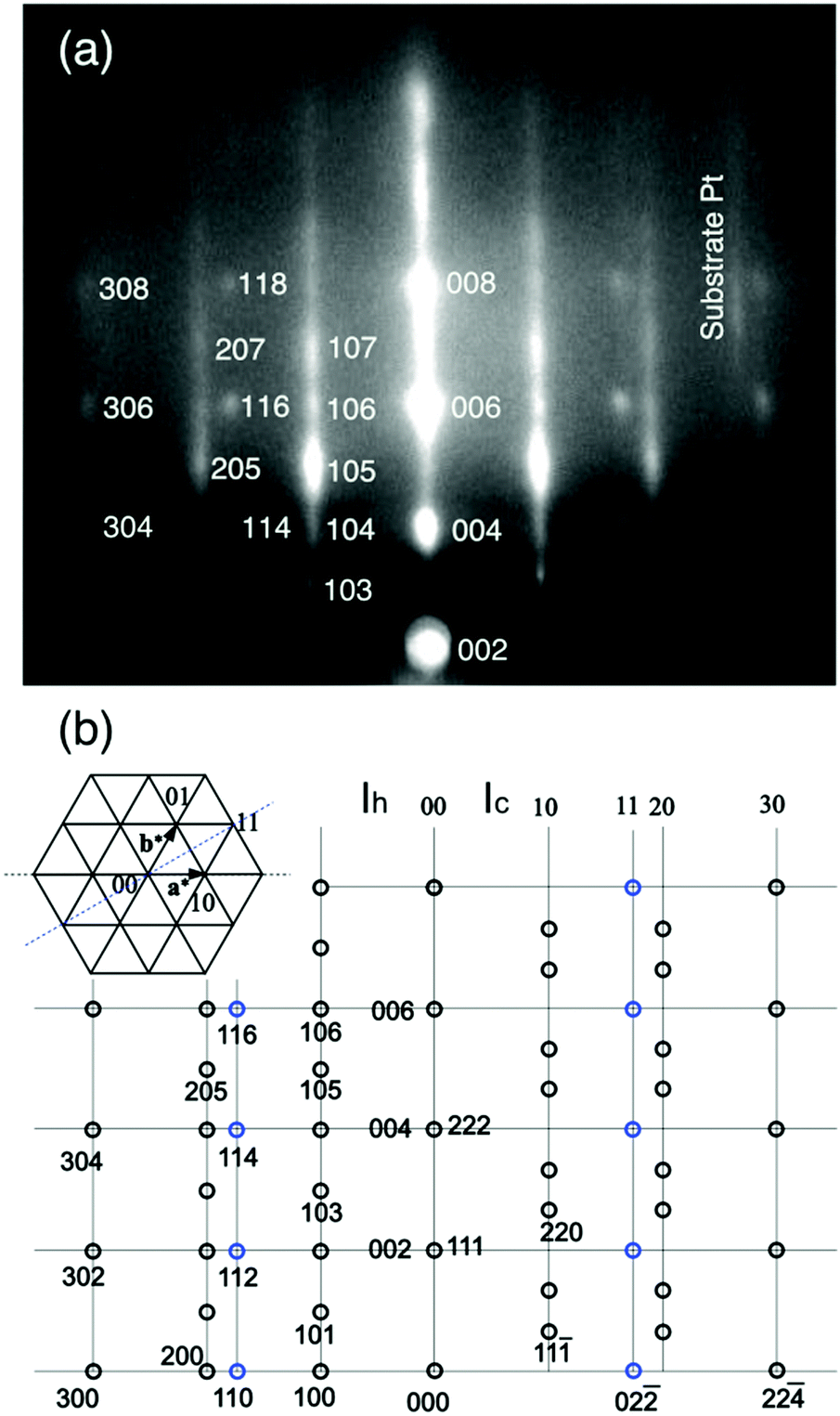 | ||
| Fig. 3 (a) The diffraction spots for the RHEED pattern in Fig. 2(d) are indexed. (b) The simulated RHEED patterns for the epitaxial Ih (left half) and Ic (right half) ice crystallites. The ices Ih and Ic are distinguishable because they give distinct spots on the 10 and 20 rods. | ||
Fig. 4 shows (a) temperature-programmed TOF-SIMS intensities and (b) TPD spectra obtained using the ASW film (2 L) formed on an oxygen-adsorbed Pt(111) substrate at 100 K. For oxygenation, the Pt(111) substrate was exposed to 5 L O2 molecules at 100 K. Most of the O2 species are adsorbed molecularly under this condition as confirmed by high-resolution electron energy loss spectroscopy (HREELS).33–35 In contrast to the result obtained using the pristine Pt(111) substrate, TOF-SIMS exhibits no steps ascribable to film morphology change in the H+ and H3O+ intensities. The increase in the Pt+(H2O)2 intensity at T > 153 K results from film dewetting and/or evaporation of water. The TPD spectrum of water exhibits a main peak at around 162 K, where the H+ and H+(H2O)2 intensities also decay in TOF-SIMS. The intact O2 molecules desorb together with water. The O2 TPD peak appears at higher temperature than that from O2 adsorbed Pt(111) (∼140 K),35 suggesting the occurrence of some attractive interaction between O2 and water. The O2 peak is considerably small, suggesting that the chemisorbed O species are formed on the surface during the heating process. The H3O+ and Pt+(H2O)2 ions remain up to 200 K where a small peak appears in water TPD, signifying the presence of water residues. This peak corresponds to the 2D peak observed at 170 K using the pristine Pt(111) substrate, but it appears at higher temperature because water molecules are bound tightly to the chemisorbed oxygen species.36 A small shoulder in the water TPD spectrum at around 157 K is ascribable to water crystallization.16,18
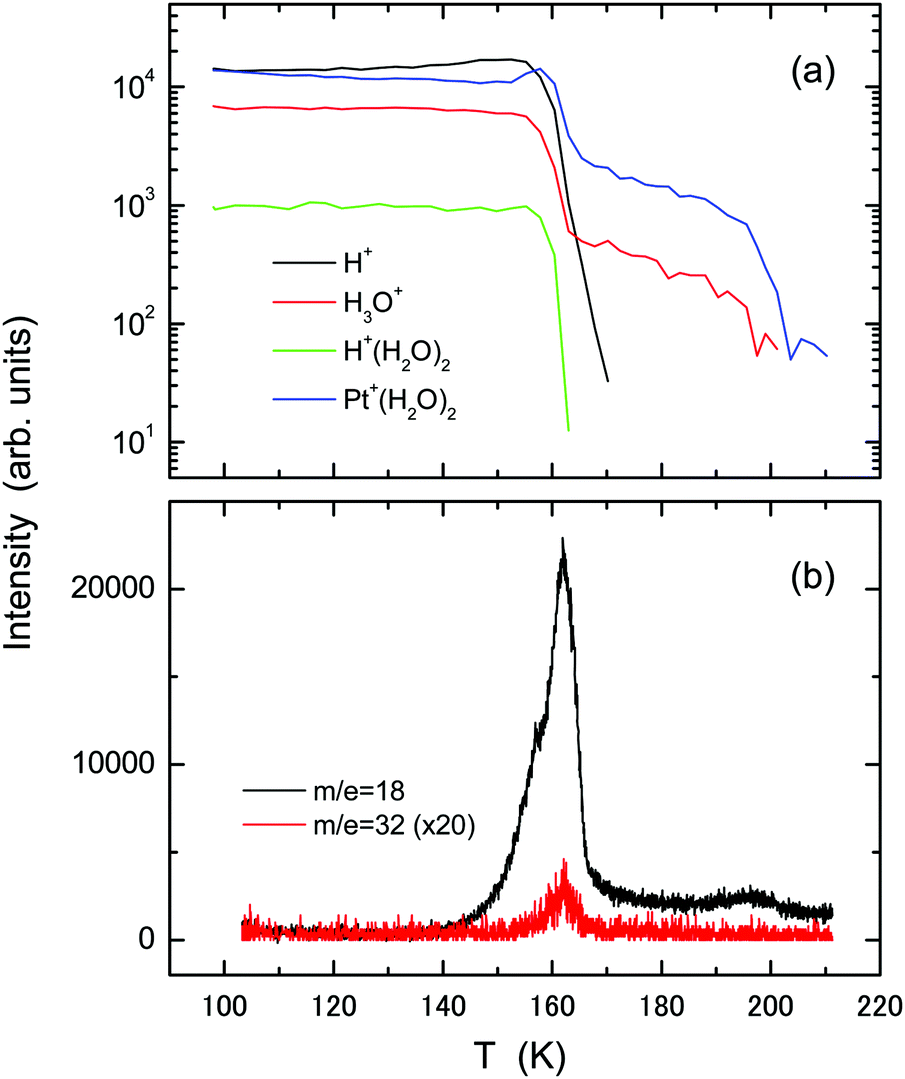 | ||
| Fig. 4 The same as in Fig. 1, but using the oxygenated Pt(111) substrate prepared by exposure to 5 L O2 molecules at 100 K. | ||
The influence of oxygen adspecies on the crystallization kinetics of ASW is also investigated based on the diffraction pattern. Fig. 5 presents RHEED images obtained during the heating process of the ASW film prepared in the same way as in Fig. 4. The RHEED image obtained by deposition of 5 L O2 molecules at 100 K (a) is almost unchanged relative to that using the bare Pt(111) substrate. The diffuse RHEED pattern of ASW appears after exposure to 2 L water (b). The pattern is fundamentally unchanged up to 150 K (c). The crystallites are formed at around 160 K (d), as confirmed from the occurrence of Bragg spots. It is revealed from the diffraction pattern that ice Ic is formed preferentially although the spots are rather vague in comparison with those of ice Ih formed on the pristine Pt(111) substrate in Fig. 2. Additional spots between the original spots of ice Ic are ascribable to the formation of twins, and a weak Debye–Scherrer ring is also identifiable. The (√3 × √3)R30° structure formed at 180 K after evaporation of crystallites (e), indicating that water residues tend to be ordered on oxygenated Pt(111). The RHEED pattern changes to a weak 2 × 2 structure along with the desorption of residual water at 200 K (f) because the chemisorbed oxygen finally forms the ordered structure.33 The crystallization kinetics of ASW is fundamentally unchanged when the film is deposited onto the substrate exhibiting the (√3 × √3)R30° pattern (not shown), suggesting that the presence of chemisorbed oxygen rather than its initial structure plays a role in controlling the epitaxial growth of the crystalline ice.
 | ||
| Fig. 5 (a) The RHEED image obtained using the oxygenated Pt(111) substrate. The ASW film was formed on it by deposition of 2 L water at 100 K; images obtained during the heating process at (b) 100, (c) 150, (d) 160, (e) 180, and (f) 200 K are displayed. | ||
Fig. 6 presents experimental results of (a) TOF-SIMS and (b) TPD obtained using the ASW film (2 L) deposited at 100 K onto the Pt(111)–CO substrate which is formed by adsorption of 5 L CO at 150 K. With increasing temperature, intensities of the H+ and Pt+(H2O)2 ions decrease gradually. The intensity of H+(H2O)2 rather increases relative to H3O+ during desorption of water molecules at T = 160–165 K. No independent TPD peak from the 2D water layer is identifiable, in agreement with decay of the Pt+(H2O)2 intensity, but a small tail exists up to 180 K, corresponding to the secondary ions from residues. The results contrast significantly to those using the pristine and oxygenated Pt(111) substrates.
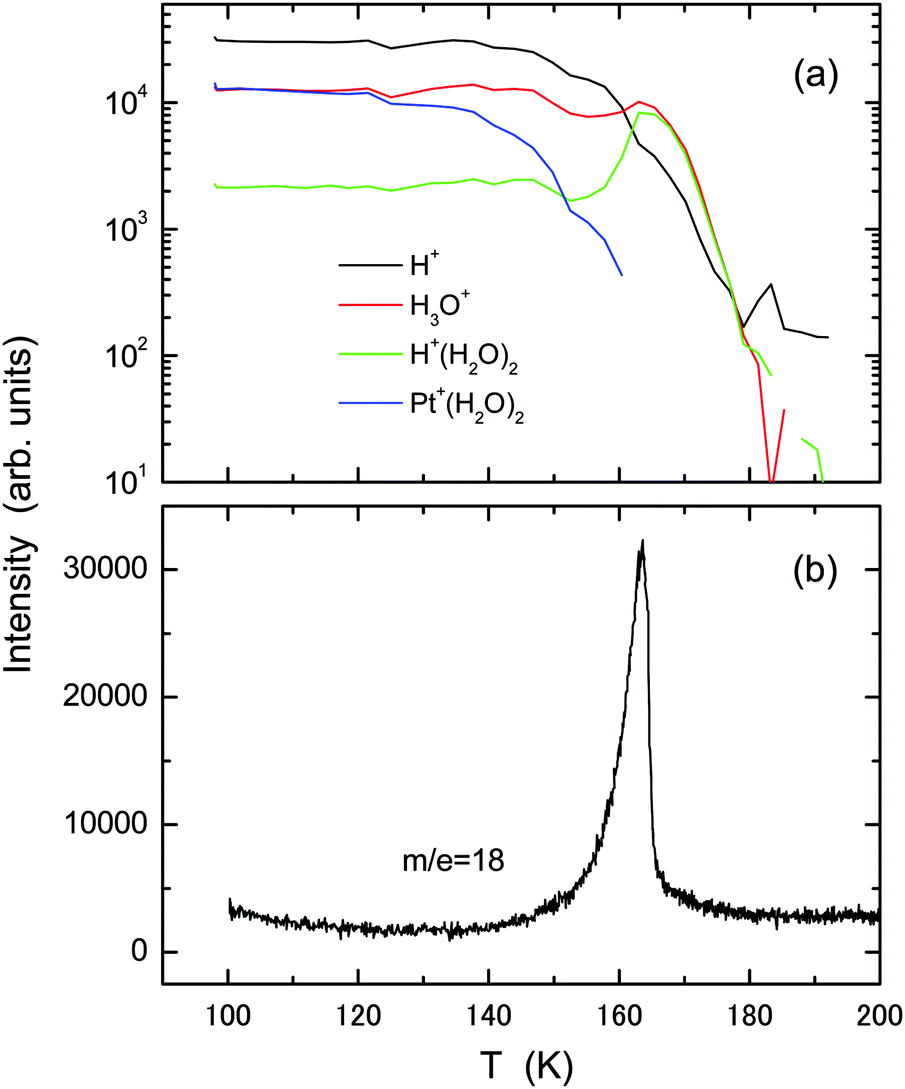 | ||
| Fig. 6 The same as in Fig. 1, but using the CO-adsorbed Pt(111) substrate prepared by exposure to 5 L CO molecules at 150 K. | ||
The temperature-programmed RHEED images obtained using the ASW film (2 L) formed on the CO-adsorbed Pt(111) substrate are shown in Fig. 7. Additional streaks appear upon CO deposition (a). CO adsorbs molecularly on Pt(111),37,38 although complicated structures are known to occur depending on its coverage. The adsorption sites of CO are still controversial between experiment and theory.38 In this study, therefore, no detailed structure of CO-adsorbed Pt(111) is discussed. The diffuse diffraction pattern of the ASW film (b) is different from the spotty patterns of crystallites at 150 K (c) and 155 K (d). The diffraction spots are streaky because of the coexistence of Ic and Ih crystallites or the formation of a stacking disordered ice.32 The sharp spots inside the second rods come from crystallites with other orientations. The Bragg spots are weakened considerably at 160 K (e), and no diffraction spots from adsorbed water are recognizable at 165 K (f) after evaporation of crystallites (see the TPD spectrum in Fig. 6(b)). However, even after evaporation of ice crystallites, the H3O+ and H+(H2O)2 ions have considerable intensity at 165 K (Fig. 6(a)), suggesting that water residues adsorb disorderly on the CO-adsorbed Pt(111) surface, or less probably, the CO-water composite layer provides a commensurate lattice to the substrate.
 | ||
| Fig. 7 (a) The RHEED image obtained using the CO-adsorbed Pt(111) substrate. The ASW film was formed on it by deposition of 2 L water at 100 K; images obtained during the heating process at (b) 100, (c) 150, (d) 155, (e) 160, and (f) 165 K are displayed. | ||
In the above context, water crystallization has been explored by heating the initial ASW films. The crystalline ice can be formed via vapor deposition when the surface diffusivity of water is kept sufficiently high. RHEED patterns obtained during water deposition at 135 K (close to the water's glass transition temperature of Tg = 136 K)17,39 are shown in Fig. 8. When the pristine Pt(111) substrate is used, streaky diffraction patterns of ice Ih are obtained after exposure to (a) 2 and (b) 20 L water. No additional spots from crystallites with other orientations are present in contrast to those descended from ASW (Fig. 2). It is known that Debye–Scherrer ring patterns are observed when thicker ASW films (15–20 L) are crystallized because randomly oriented crystallites are formed.24,29 However, ordered crystallites continue to grow till considerably high exposures during high-temperature water deposition because the ice is grown homoepitaxially on smaller grains (i.e., nuclei) formed initially. It is thus suggested that the epitaxial growth occurs more preferentially during slow water–vapor deposition at around Tg rather than the nucleation and growth process of the initial ASW films occurring at around 150 K. The RHEED patterns in Fig. 8(c and d), respectively, are obtained during deposition of 2 and 20 L water molecules onto the oxygenated Pt(111) substrate that initially exhibited the (√3 × √3)R30° pattern. In this case, the ice Ic crystallites are formed predominantly. The Debye–Scherrer ring pattern is weakened in comparison with that in Fig. 5(d) even for the thicker film. However, crystallinity of the film is apparently poor compared to that formed on the pristine Pt(111) substrate. During water vapor deposition on the CO-adsorbed Pt(111) substrate, the ice Ic crystallite is grown initially at exposure to 2 L water (e), which is followed by the growth of ice Ih after exposure to 20 L (f). The RHEED patterns of ice formed on the CO-adsorbed Pt(111) substrate (e and f) are spottier than those obtained using the pristine Pt(111) substrate (a and b), suggesting the growth of larger 3D crystallites. Consequently, the effect of CO adspecies is apparently weak relative to that of chemisorbed oxygen in terms of the epitaxial growth of crystalline ices on Pt(111).
 | ||
| Fig. 8 RHEED images of crystalline ices formed by water vapor deposition onto the pristine (a and b), oxygenated (c and d), and CO-adsorbed (e and f) Pt(111) substrates at 135 K. The mages obtained by water exposure to 2 L (a, c and e) are compared with those of 20 L (b, d and f). | ||
4 Discussion
The crystallization kinetics of thin ASW films, as well as the water–Pt interactions, are influenced strongly by the oxygen adspecies on the Pt(111) substrate. The nucleation temperature of water becomes higher (155–160 K), as revealed from both the RHEED pattern and film morphology change. Probably, reorganization of water molecules to create nuclei at the substrate interface is disturbed when the hydrogen bonds are formed with the chemisorbed oxygen. Consequently, the water crystallization temperature approaches to the spontaneous nucleation temperature of bulk water (∼160 K)39 using the oxygenated Pt(111) substrate. The ice Ic is formed preferentially instead of ice Ih with the epitaxial relationship of (111)Ic//(111)Pt and [1![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 0]Ic//[11]Pt. Along with this behavior, the characteristic shoulder during crystallization becomes identifiable in the water TPD spectrum. The desorption rate of water molecules is associated with the film morphology change, so that the TPD shoulder might be hindered when the crystal grains are created gradually via the substrate induced nucleation at lower temperatures than the spontaneous nucleation. After evaporation of crystallites, the 2D layer remains on the oxygenated Pt(111) surface till higher temperature (∼200 K) than using the pristine Pt(111) substrate. This behavior is ascribable either to the formation of hydrogen bonds between water and chemisorbed oxygen atoms or to the dissociative adsorption of water molecules to form hydroxyl groups. The former is not sufficient to make water residues adsorb up to 200 K, as evidenced by the result for water adsorption on a hydroxylated Si substrate.18 It is well established that water is dissociated by chemisorbed oxygen on Pt(111).1,2 However, the H3O+ and Pt+(H2O)2 ions are emitted from the 2D layer, which suggests that water residues are not dissociated completely. The experimental result using the oxygenated Pt(111) substrate resembles that of the water monolayer formed on the pristine Ru(0001) substrate, where water molecules are partially dissociated.10 The strong suppression of the H+ yield relative to H3O+ in Fig. 4(a) is explainable as that the hydroxyl group of water tends to be directed towards the substrate or parallel to the surface via the formation of hydrogen bonds with the chemisorbed oxygen atoms. The partially dissociated water species are pinned at the chemisorbed oxygen sites, thereby formation of the (√3 × √3)R30° structure for the 2D layer on the oxygenated Pt(111) substrate. In contrast, the 2D layer of intact water formed on the pristine Pt(111) substrate is not ordered at higher temperatures because of the absence of hydrogen bonding with the substrate. The water molecule in direct contact with the substrate behaves like a liquid that is equilibrated with the premelting layer of crystallites.28,29,40 The liquidlike 2D layer is expected to be ordered by cooling. Probably, such water species form hexagonal rings both in registry and out-of-registry with the substrate atoms, as imaged by STM at lower temperatures.6,41
0]Ic//[11]Pt. Along with this behavior, the characteristic shoulder during crystallization becomes identifiable in the water TPD spectrum. The desorption rate of water molecules is associated with the film morphology change, so that the TPD shoulder might be hindered when the crystal grains are created gradually via the substrate induced nucleation at lower temperatures than the spontaneous nucleation. After evaporation of crystallites, the 2D layer remains on the oxygenated Pt(111) surface till higher temperature (∼200 K) than using the pristine Pt(111) substrate. This behavior is ascribable either to the formation of hydrogen bonds between water and chemisorbed oxygen atoms or to the dissociative adsorption of water molecules to form hydroxyl groups. The former is not sufficient to make water residues adsorb up to 200 K, as evidenced by the result for water adsorption on a hydroxylated Si substrate.18 It is well established that water is dissociated by chemisorbed oxygen on Pt(111).1,2 However, the H3O+ and Pt+(H2O)2 ions are emitted from the 2D layer, which suggests that water residues are not dissociated completely. The experimental result using the oxygenated Pt(111) substrate resembles that of the water monolayer formed on the pristine Ru(0001) substrate, where water molecules are partially dissociated.10 The strong suppression of the H+ yield relative to H3O+ in Fig. 4(a) is explainable as that the hydroxyl group of water tends to be directed towards the substrate or parallel to the surface via the formation of hydrogen bonds with the chemisorbed oxygen atoms. The partially dissociated water species are pinned at the chemisorbed oxygen sites, thereby formation of the (√3 × √3)R30° structure for the 2D layer on the oxygenated Pt(111) substrate. In contrast, the 2D layer of intact water formed on the pristine Pt(111) substrate is not ordered at higher temperatures because of the absence of hydrogen bonding with the substrate. The water molecule in direct contact with the substrate behaves like a liquid that is equilibrated with the premelting layer of crystallites.28,29,40 The liquidlike 2D layer is expected to be ordered by cooling. Probably, such water species form hexagonal rings both in registry and out-of-registry with the substrate atoms, as imaged by STM at lower temperatures.6,41
The nucleation temperature of water on CO-adsorbed Pt(111) is basically the same as that on pristine Pt(111). The lack of the shoulder in the water TPD spectrum is also consistent with this behavior. The RHEED pattern from crystallites becomes spottier than that using pristine Pt(111), and stacking disorders are formed considerably. It should be noticed that the H+(H2O)2 ions increase in intensity after crystallization occurs at ca. 150 K. The enhancement of cluster ion intensities during sputtering has been reported for water adsorbed on a hydrogenated Pt(111) substrate,42 but the present result has nothing to do with this phenomenon. The decrease in the Pt+(H2O)2 intensity at T > 130 K in Fig. 6(a) strongly suggest that water molecules tend to interact with CO on the Pt(111) substrate along with weakening of the water–Pt interaction at higher temperatures. Consequently, the Pt(111) substrate becomes hydrophobic upon adsorption of CO molecules. It is known that cluster ions, H+(H2O)n, are emitted efficiently via an electronic sputtering process when water is adsorbed on molecular solid films because of the localization of ionic holes or the occurrence of “Coulomb explosion” of water nanoclusters.43 The smaller the cluster size is, the more efficiently cluster ions are emitted. The same is expected to occur when “potential sputtering” occurs using the primary He+ ions.44 Probably, this is a reason why the H+(H2O)2 ion increases in intensity after evaporation of crystallites at 162 K (Fig. 6). It is also possible that the CO adspecies undergoes “hydrophobic hydration”. The fact that the H+ intensity decreases relative to the H3O+ intensity at T > 160 K might be ascribed to the formation of hydration shells of water whose OH group is direct towards the hydrophobic CO solute. Such a structure has been predicted to occur during interactions of water with hydrophobic species based on molecular dynamics simulations.45–47 Probably, this provides a reason why the water-CO interactions are attractive when water is physisorbed on a saturated CO monolayer on Pt(111).2 Consequently, the epitaxial growth of crystallites is realized more preferentially in the presence of the CO molecule rather than the chemisorbed oxygen because the former does not disturb the reorganization of hydrogen bonds during water nucleation at the substrate interface.
5 Conclusions
How the water molecules in the monolayer regime interact with oxygen and carbon-monoxide adspecies on Pt(111) was investigated in comparison with the results using the pristine Pt(111) substrate in terms of the crystallization kinetics and surface wettability. The ASW film on the Pt(111) surface crystallizes above 150 K, thereby forming epitaxially-grown ice Ih grains and a disordered 2D layer. The latter survives up to 175 K after evaporation of crystallites at 165 K. The ASW film crystallizes into ice Ic preferentially on the oxygenated Pt(111) substrate, where ordering of the crystallites with respect to the substrate is poor in comparison with that on the pristine Pt(111) substrate. The nucleation temperature increases as well because reorganization of water molecules to form nuclei is disturbed by hydrogen bonds with the chemisorbed oxygen. Such water species are dissociated partially and exhibit the (√3 × √3)R30° structure after disappearance of crystallites up to 200 K. The stacking disordered ice or a mixture of Ic and Ih crystallites is formed on the CO-adsorbed Pt(111) substrate when ASW is heated to 150 K. This substrate appears to be hydrophobic because the water–Pt contact is hampered with increasing temperature. The yield of water cluster ions increases during and after evaporation of crystallites, indicating that water residues interacting preferentially with the CO adspecies are sputtered efficiently because of the isolation from the metal substrate.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was partly funded by the Japan Society for the Promotion of Science through a Grant-in-Aid for Scientific Research (C), No. 22540339.References
- P. A. Thiel and T. T. Madey, Surf. Sci. Rep., 1987, 7, 211 CrossRef CAS.
- M. A. Henderson, Surf. Sci. Rep., 2002, 46, 1 CrossRef CAS.
- H. Ogasawara, B. Brena, D. Nordlund, M. Nyberg, A. Pelmenschikov, L. G. M. Petterrsson and A. Nilsson, Phys. Rev. Lett., 2002, 89, 276102 CrossRef CAS PubMed.
- A. Glebov, A. P. Graham, A. Menzel and J. P. Toennies, J. Chem. Phys., 1997, 106, 9382 CrossRef CAS.
- S. Haq, J. Harnett and A. Hodgson, Surf. Sci., 2002, 505, 171 CrossRef CAS.
- S. Nie, P. J. Feibelman, N. C. Bartelt and K. Thrumer, Phys. Rev. Lett., 2010, 105, 026102 CrossRef CAS PubMed.
- A. Hodgson and S. Haq, Surf. Sci. Rep., 2009, 64, 381 CrossRef CAS.
- Y. Lilach, M. J. Iedama and J. P. Cowin, Phys. Rev. Lett., 2007, 98, 016105 CrossRef PubMed.
- C. Clay, S. Haq and A. Hodgson, Phys. Rev. Lett., 2004, 92, 046102 CrossRef CAS PubMed.
- P. J. Feibelman, Science, 2002, 295, 99 CrossRef CAS PubMed.
- N. Materer, U. Starke, A. Barbieri, M. A. Van Hove, G. A. Somorjay, G.-J. Kroes and C. Minot, J. Phys. Chem., 1995, 99, 6267 CrossRef CAS.
- R. S. Smith, C. Huang, E. K. L. Wong and B. D. Kay, Surf. Sci., 1996, 367, L13 CrossRef CAS.
- P. Lofgren, P. Ahlstrom, D. V. Chakarov, J. Lausmaa and B. Kasemo, Surf. Sci., 1996, 367, L19 CrossRef.
- P. Lofgren, P. Ahlstrom, J. Lausma, B. Kasemo and D. Chakarov, Langmuir, 2003, 19, 265 CrossRef.
- S. M. McClure, E. T. Barlow, M. C. Akin, D. J. Safarik, T. M. Truskett and C. B. Mullins, J. Phys. Chem. B, 2006, 110, 17987 CrossRef CAS PubMed.
- R. S. Smith, C. Huang, E. K. L. Wong and B. D. Kay, Phys. Rev. Lett., 1997, 79, 909 CrossRef CAS.
- R. S. Smith, C. Huang and B. D. Kay, J. Phys. Chem. B, 1997, 101, 6123 CrossRef CAS.
- R. Souda, J. Phys. Chem. C, 2012, 116, 20895 CrossRef CAS.
- A. S. Bolina, A. J. Wolff and W. A. Brown, J. Phys. Chem. B, 2005, 109, 16836 CrossRef CAS PubMed.
- J. Günster and R. Souda, Langmuir, 2006, 22, 6939 CrossRef PubMed.
- C.-Y. Ruan, V. A. Lobastov, F. Vigliotti, S. Chen and A. H. Zewail, Science, 2004, 304, 80 CrossRef CAS PubMed.
- D.-S. Yang and A. H. Zewail, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 4122 CrossRef CAS PubMed.
- X. He, C. Wu and D.-S. Yang, J. Chem. Phys., 2016, 145, 171102 CrossRef PubMed.
- R. Souda and T. Aizawa, Phys. Chem. Chem. Phys., 2018, 20, 21856 RSC.
- P. Jenniskens, S. F. Banham, D. F. Blake and M. R. S. McCoustra, J. Chem. Phys., 1997, 107, 1232 CrossRef CAS PubMed.
- E. H. G. Backus, M. L. Grecea, A. W. Kleyn and M. Bonn, Phys. Rev. Lett., 2004, 92, 236101 CrossRef PubMed.
- R. Souda, Phys. Rev. Lett., 2004, 93, 235502 CrossRef PubMed.
- R. Souda, J. Phys. Chem. C, 2017, 121, 12199 CrossRef CAS.
- R. Souda and T. Aizawa, J. Phys. Chem. C, 2018, 122, 28094 CrossRef CAS.
- S. Haq, J. Harnett and A. Hodgson, Surf. Sci., 2002, 505, 171 CrossRef CAS.
- G. A. Kimmel, N. G. Petrik, Z. Dohnalek and B. D. Kay, Phys. Rev. Lett., 2005, 95, 166102 CrossRef PubMed.
- T. L. Malkin, B. J. Murray, A. V. Brukhno, J. Anwar and C. G. Salzmann, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 1041 CrossRef CAS PubMed.
- J. L. Gland and B. A. Sexton, Surf. Sci., 1980, 95, 587 CrossRef CAS.
- H. Steininger, S. Lehwald and H. Ibach, Surf. Sci., 1982, 123, 1 CrossRef CAS.
- N. R. Avery, Chem. Phys. Lett., 1983, 96, 371 CrossRef CAS.
- G. Zimbitas, M. E. Gallaghher, G. R. Darling and A. Hodgson, J. Chem. Phys., 2008, 128, 074701 CrossRef PubMed.
- H. Steininger, S. Lehwald and H. Ibach, Surf. Sci., 1982, 123, 264 CrossRef CAS.
- P. J. Feibelman, B. Hammer, J. K. Norskov, F. Wagner, M. Scheffler, R. Stumpf, R. Watwe and J. Dumesic, J. Phys. Chem. B, 2001, 105, 4018 CrossRef CAS.
- G. P. Johari, A. Hallbrucker and E. Mayer, Nature, 1987, 330, 552 CrossRef CAS.
- R. Souda, Phys. Chem. Chem. Phys., 2014, 16, 1095 RSC.
- S. Maier, B. A. J. Lechner, G. A. Somorjai and M. Salmeron, J. Am. Chem. Soc., 2016, 138, 3145 CrossRef CAS PubMed.
- Y. Kim, S. Shin and H. Kang, Angew. Chem., Int. Ed., 2015, 26, 7626 CrossRef PubMed.
- R. Souda, Phys. Rev. B: Condens. Matter Mater. Phys., 2002, 65, 245419 CrossRef.
- R. Souda, J. Phys. Chem. A, 2007, 111, 201 CrossRef CAS PubMed.
- C. Y. Lee, J. A. McCammon and P. J. Rossky, J. Chem. Phys., 1984, 80, 4448 CrossRef CAS.
- T. A. Ho and A. Striolo, J. Chem. Phys., 2013, 138, 054117 CrossRef PubMed.
- M. K. Rana and A. Chandra, J. Chem. Phys., 2013, 138, 204702 CrossRef PubMed.
| This journal is © the Owner Societies 2019 |