
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Quantitative analysis of 14N quadrupolar coupling using 1H detected 14N solid-state NMR†
James A.
Jarvis‡
a,
Maria
Concistre
b,
Ibraheem M.
Haies
bc,
Richard W.
Bounds
b,
Ilya
Kuprov
 b,
Marina
Carravetta
b and
Philip T. F.
Williamson
*a
b,
Marina
Carravetta
b and
Philip T. F.
Williamson
*a
aCentre for Biological Sciences, University of Southampton, SO17 1BJ, Southampton, UK. E-mail: P.T.Williamson@soton.ac.uk
bSchool of Chemistry, University of Southampton, SO17 1BJ, Southampton, UK
cDepartment of Chemistry, College of Science, University of Mosul, Mosul, Iraq
First published on 27th February 2019
Abstract
Magic-angle spinning solid-state NMR is increasingly utilized to study the naturally abundant, spin-1 nucleus 14N, providing insights into the structure and dynamics of biological and organic molecules. In particular, the characterisation of 14N sites using indirect detection has proven useful for complex molecules, where the ‘spy’ nucleus provides enhanced sensitivity and resolution. Here we exploit the sensitivity of proton detection, to indirectly characterise 14N sites using a moderate rf field to generate coherence between the 1H and 14N at moderate and fast-magic-angle spinning frequencies. Efficient numerical simulations have been developed that have allowed us to quantitatively analyse the resulting 14N lineshapes to determine both the size and asymmetry of the quadrupolar interaction. Exploiting only naturally occurring abundant isotopes will aid the analysis of materials with the need to resort to isotope labelling, whilst providing additional insights into the structure and dynamics that the characterisation of the quadrupolar interaction affords.
Introduction
Nitrogen-14 has a natural abundance of >99.6% and a moderate gyromagnetic ratio (γ14N/γ1H ∼ 0.07), making it a potentially attractive nucleus for high-resolution solid-state NMR studies of nitrogen containing compounds. However, 14N is a spin-1 nucleus with a nuclear quadrupole interaction (NQI) typically on the order of 1–5 MHz, making detection challenging. The NQI is however sensitive to variations in molecular symmetry, geometry and electrostatic environment, which in combination with its high natural abundance and prevalence in organic and biological materials, makes the development of methods to characterize 14N sites desirable. To this end, a number of methods have been presented in the literature, particularly within the last ten years, that facilitate the determination of NQI parameters of nitrogen sites in a range of organic, biological and pharmaceutical materials.Despite the size of the NQI and the width of resulting signal, ultra-wideline excitation methods developed by Schurko and co-workers have provided accurate NQI parameters at 14N sites in a number of organic and pharmaceutical compounds.1–3 The resolution of this method is inherently limited by the width of static 14N signals, which overlap and hinder interpretation, limiting this technique to compounds with few (currently fewer than 3) unique 14N sites.
Methods for detection of overtone (Δm = ±2) transitions on static samples,4–6 and more recently under MAS7–12 and DOR13 have provided alternative methods for characterizing 14N sites. Unlike Δm = ±1 14N signals, overtone signals are not affected by the large (MHz) quadrupole broadening. However, there are sensitivity concerns when exciting this “forbidden” transition, which are further exacerbated by the slow nutation and correspondingly low excitation bandwidth of overtone signals, making simultaneous detection of multiple 14N overtone signals difficult.8
A third strategy for detection of 14N is the indirect detection method, pioneered independently by Gan and Bodenhausen's laboratories,14–25 where 14N sites are detected via their interaction with coupled spin-1/2 ‘spy’ nuclei; typically 1H or 13C, using pulse sequences qualitatively similar to HMQC experiments. Transfer of polarisation between the 14N and the ‘spy’ nucleus occurs due to high-order cross-terms between the quadrupolar and 14N‘spy’ dipolar Hamiltonians, known as the ‘residual dipolar splitting’ (RDS), as well as a contribution from the J-coupling, in experiments known as J-HMQCs. Alternatively, polarisation transfer can be driven by the recoupling of the 1H/14N dipolar coupling using rotary resonance (R3), REDOR or symmetry based recoupling sequences, in sequences that have been generally termed D-HMQC experiments.19,26,27 Similarly, double cross-polarization schemes have also been proposed for indirect detection which function well at higher spinning speeds.28 These indirect techniques benefit from the sensitivity and resolution that are afforded by the ‘spy’ nuclei and do not require excitation or detection of the entire first-order broadened 14N spectrum. Accordingly, they have found application to the study of structure and dynamics of a number of systems.18,29,30 One of the major obstacles to the more widespread application of these techniques is their relatively poor spy nucleus/14N transfer efficiencies, which usually result in a ∼90% drop in sensitivity compared to the 1H spin-echo, and the strong dependency on high 14N RF amplitudes,31 this is particularly acute at moderate spinning speeds (<60 kHz). This, in turn, renders the accurate de novo determination of 14N NQI parameters from these experiments difficult, since precise determination of the quadrupolar coupling constant (CQ) and asymmetry parameter (η) requires fitting of the 14N lineshape, which cannot be achieved without high quality spectra with a high S/N ratio.
In this study we present a method for indirect detection of 14N signals via1H's with enhanced efficiency at moderate to fast MAS frequencies (35–70 kHz), with transfer efficiencies of up to 27.5%, enabling acquisition of 1H/14N correlation spectra with improved signal/noise ratios and well-defined lineshapes in the 14N dimension. This method, which is derived from our earlier work on the detection of 14N via13C,32 differs from previously published methods of indirect 14N detection based on the HMQC pulse sequence, in that polarisation transfer occurs not under free evolution or established methods of heteronuclear dipolar recoupling, but rather under relatively long (100s μs) periods of moderate (30–70 kHz) RF irradiation on the 14N channel (see Fig. 1). As described in our earlier studies, during these periods of irradiation both single and double quantum coherences are generated with efficiencies in excess of 20% of the initial equilibrium magnetization.32 As per previous indirect detection sequences the indirect 14N signals in 2D spectra are not affected by the typically large (MHz) first order quadrupolar interaction providing that the t1 increments are rotor synchronised and the magic-angle accurately set. This results in 14N signals at a frequency which is the sum of the chemical shift and second order isotropic quadrupolar shift (δisoQ), given for the general case in ppm by:29
 | (1) |
 | (2) |
 | (3) |
 | ||
| Fig. 1 Pulse sequences employed for the acquisition of the 2D-14N/1H (A) and 3D-14N/1H/1H correlation (B) spectra reported in this study. The length of the excitation (τexc) and reconversion pulses (τrec) and the t1 increments are chosen to ensure that the sequence is rotor synchronised, π/2 and π pulses are highlighted in black and grey respectively. For the 3D experiment, proton mixing during τm was conducted using a RFDR pulse train, pulses have been omitted for clarity. | ||
The simulation of indirectly detected 14N lineshapes is difficult since the lineshape depends on numerous parameters, including quadrupolar broadening, and the orientation of the quadrupolar tensor with respect to the dipolar and CSA tensors. Therefore, determination of 14N NQI parameters has not been previously attempted due to (a) poor efficiency of indirect detection methods leading to noisy and ill-defined 14N lineshapes unsuitable for fitting, and (b) computationally expensive nature of simulating these experiments. In this work, we address both of these issues and present a highly efficient 1H/14N correlation experiment, and fast and accurate simulations which in combination allow the de novo determination of 14N NQI parameters from organic solids with a variety of 14N sites with various NQI parameters. These fast and accurate numerical simulations of each 14N site allows the fitting of simulations to experimental lineshapes and the simultaneous determination of 14N NQI parameters at multiple sites from a 2D data set.
Materials and methods
Materials
Samples of histidine hydrochloride monohydrate (His·HCl·H2O), L-histidine (L-His), N-acetyl-valine (NAV) and valine were purchased from Sigma-Aldrich (UK) and used without further purification.Solid-state NMR experiments
Solid-state NMR experiments on His·HCl·H2O and L-His were performed on an Agilent DDR2 spectrometer operating at 14.1 T (Larmor frequencies of 600 MHz and 43.4 MHz for 1H and 14N) equipped with a 1.6 mm triple resonance MAS probe tuned in double resonance mode. MAS frequencies were 40 kHz for the L-His sample and 35.5 kHz for the His·HCl·H2O sample.The pulse sequence for indirect detection of 14N via1H used in this work is shown in Fig. 1. Pulse lengths for 1H pulses were π/2 = 1.55 μs, π = 3.10 μs on both samples, 14N pulses for excitation and reconversion were applied at 62.5 kHz for 575 μs, and 30 kHz for 390 μs for L-His and His·HCl·H2O, respectively. The L-His spectrum was recorded with 112 scans per t1 increment, with 160 complex increments and a recycle delay of 2 s and the His·HCl·H2O spectrum was recorded with 64 scans and 254 t1 increments with a recycle delay of 2 s. Both were recorded using States-TPPI.33 Both datasets were zero-filled to 2048 × 2048 points and 2D Fourier transformed without a window function.
Experiments on NAV were performed on a Bruker Avance II spectrometer at 20 T (Larmor frequencies of 850 MHz and 61.4 MHz for 1H and 14N) using a JEOL 1 mm double resonance MAS probe. The sample was spun with a MAS frequency of 78 kHz. Spectra were acquired using the pulse sequence in Fig. 1. 1H pulses were π/2 = 1.1 μs, π = 2.2 μs and 14N excitation and reconversion pulses were applied at 72 kHz for 370 μs. 128 complex t1 increments were recorded with 136 scans per increment, in a phase sensitive manner, using States-TPPI.33 The recycle delay was 3 s. Data was zero-filled to 2048 points in each dimension before 2D Fourier transform. 3D 14N resolved 1H/1H correlation spectra were acquired the pulse scheme shown in Fig. 1B. Following 14N evolution, protons were allowed to evolve under the proton chemical shift prior a period of RFDR proton/proton mixing34 prior to detection. Data was acquired with States-TPPI33 in both indirect dimensions with 16 and 32 increments in the indirect 14N and 1H dimensions respectively. Data was processed in nmrPipe35 and visualised in Analysis 2.4.0, modified to include 14N parameters.36 In all spectra, the 14N dimension is referenced to the 14N resonance of NH4Cl at 39.3 ppm, ensuring that the 14N shifts can be compared directly with values on the scale typically used for referencing 15N in biological solids.37
Numerical simulations
All NMR simulations were performed with the Spinach library versions 1.6 and 2.2.38 Each nitrogen site simulated was approximated as a pair of nuclei comprising the 14N nucleus and the closest bound proton. Simulations included dipolar coupling, CSA and NQI parameters for both spins that were initially obtained from CASTEP calculations39 or literature values. These initial calculated and literature values are shown in Table 1. Relative orientations of 14N NQI and 14N and 1H CSA tensors were also obtained from the literature, and are also given in Table 1, defined with respect to the PAS of the 1H–14N dipolar tensor. Relaxation was neglected in the simulations, and an apodization function applied to simulated FIDs before Fourier transform that reflected the effective T2 of the experimental 14N signal. An input script for the experiment described in this paper is available in the latest release of the Spinach library. For simulations on sites in L-His and His·HCl·H2O, powder averaging was performed over 770 pairs of α and β angle orientations calculated using the Lebedev method. A Floquet rank of 45 was sufficient for convergence for all sites in L-His and His·HCl·H2O except the Nδ site of His·HCl·H2O, where a rank of 55 was required due to the increased CQ magnitude. Simulations of NAV used 1454 α and β angle orientations and a Floquet rank of 55. To fit the experimentally determined lineshapes the simulated 14N signals, CQ and η were systematically varied in steps of 50 kHz and 0.1 respectively and the RMSD between experiment and simulation calculated.| Sample | Site | δ (ppm) | C Q (MHz) | η | Orientation (α, β, γ) | ||
|---|---|---|---|---|---|---|---|
| 14N CQ | 14N CSA | 1H CSA | |||||
| a Measured on 15N using CPMAS. b Ref. 7. c Calculated values using CSD reference code: HISTCM12. d Ref. 43 and 44. As referenced to NH4Cl at 39.3 ppm. e Ref. 45–47. As referenced to NH4Cl at 39.3 ppm. f Ref. 48. g Ref. 49. h Ref. 50. | |||||||
| His·HCl·H2O | NH3+ | 47.3a,d | 1.32c | 0.06 | 0, 0, 0 | — | 109, 72, 172 |
| Nδ | 189.8a,d | 1.53c | 0.23 | 0, 90, 0 | −71.18, 1.5, −7.2 | 102.1, 66, −84 | |
| Nε | 177.2a,d | 1.33c | 0.94 | −75, 181.5, −7.2 | 90, 196, −86 | ||
| L-His | NH3+ | 41.6a | 1.22b | 0.15 | 0, 0, 0 | — | 109, 72, 172 |
| Nε | 168.9a | 1.44 | 0.92 | 0, 90, 0 | 75, 181.5, −7.2 | 90, 196, −86 | |
| NAV | N1 | 127.7 | 3.21f | 0.32 | 13.2, 90, 103g | 51, 20, 0e | 103.2, 0, 193h |
CASTEP calculations
To calculate the NQI we used the CASTEP density function theory (DFT) package39–42 which uses the gauge including projector augmented wave (GIPAW) algorithm. We used the Perdew–Burke–Ernzerhof (PBE) generalised gradient approximation (GGA) with ultrasoft pseudopotentials and 0.1k points Å3. We tested the value of the NQI by converging against the plane-wave energy cut-off tolerance.Results and discussion
Histidine hydrochloride monohydrate and L-histidine
In order to assess the effects of alternate hydrogen bonding patterns and changes in local geometry on 14N NQI parameters at a number of different nitrogen sites, and how these changes affect 14N lineshapes and peak positions, we initially investigated samples of L-His and His·HCl·H2O. These two salts of histidine exhibit different protonation states (see Fig. 2) and H-bonding networks, resulting in a variation in local electronic structure with the associated variation in 14N NQI parameters (Table 1). | ||
| Fig. 2 1H/14N correlation spectra of two forms of histidine at 14.1 T. Spectrum of His·HCl·H2O recorded at 35.5 kHz (A) MAS frequency L-His spectrum (B). Recorded at 40 kHz MAS frequency. Experimental (solid) and simulated (dashed) projections through the 14N dimension are plotted for each spectrum at the NH3+ site (red), the Nε site (blue) and the Nδ site (yellow). The projections are normalised to the maximum peak intensity in each respective spectrum. | ||
Fig. 2A shows the 1H/14N correlation spectrum of His·HCl·H2O, recorded at 14.1 T, under 35.5 kHz MAS. Three peaks are observed, corresponding to the three protonated nitrogen sites in this molecule. The NH3+, Nδ and Nε and sites appear at 14N shifts of 100 ppm, 263 ppm and 280 ppm, respectively. The transfer efficiency of this experiment was 27.5% at Nε, 24.4% at Nδ and 7.5% at NH3+ sites, expressed relative to a 1H spin echo on the same sample using delays equal to the length of applied 14N pulses. These efficiencies are comparable with or in excess of those reported in the literature for the same or similar compounds acquired with both J- and D-HMQC sequences. Using the J-HMQC sequence under ultrafast MAS (90 kHz), which is optimal for the 1H detected J-HMQC sequence since 1H coherence lifetimes that these experiments are particularly sensitive to increase significantly with MAS, 14N transfer efficiencies of 7.55% and 9.75% with respect to 1H spin echo were recently reported for the Nε and Nδ sites in His·HCl·H2O.51 Using a D-HMQC sequence, with R3 recoupling, transfer efficiencies of 5–6% have been reported for the NH3+ site in glycine at 20 kHz MAS.16 The experiment performed here was optimised (in terms of 14N offset and pulse length) such that efficiency was maximally distributed over the two heterocyclic 14N sites, and as such, 14N pulse widths and amplitudes are possible that increase the efficiency at any of the three sites, at the expense of signal intensity at the other sites. This results in a spectrum with a quality and signal/noise ratio such that subtle spectral features such as the shoulders on the peaks of Nε and Nδ are observed which can aid in the characterisation of the quadrupolar interaction.
Fig. 2B shows a 1H/14N correlation spectrum of L-His, recorded at 14.1 T under 40 kHz MAS. In this spectrum only two peaks are observed which are the NH3+ and Nε sites at 100 ppm and 278 ppm 14N shifts, respectively; the unprotonated Nδ site is not detected. Similar transfer efficiencies were achieved as those for His·HCl·H2O, with 20.0% and 16.6% of 1H spin echo signal recorded at Nε and NH3+ sites, respectively. This experiment was optimised for an even distribution of intensity between the two 14N sites and could be optimised to achieve a higher signal intensity at one site, potentially at the expense of that at the other.
Comparing the peaks from the Nε and NH3+ sites in L-His to the peaks from the same sites in His·HCl·H2O, there are differences in both 14N frequency and peak shape. The 14N frequency may be used to determine 14N δisoQ at each site. Contributions from the 14N chemical shift and 14N δisoQ to the observed 14N shift at each peak in the spectra of L-His and His·HCl·H2O are separated by subtracting the contribution from the isotropic chemical shifts, which are shown in Table 1. These were determined from a separate natural abundance 15N CPMAS experiment performed on each sample. For both materials, the measured values of 14N δisoQ are tabulated in Table 2.
| Sample | Site | δ isoQ expt.a (ppm) | δ isoQ calc.b (ppm) |
|---|---|---|---|
| a The centre of gravity of the 14N peak in the indirect dimension – 15N δisoCS from Table 1. A ±5 ppm error is associated with measuring the 14N frequency from the spectra. b Calculated from CQ and η from Table 1 using eqn (2), with Larmor frequencies calculated from B0 = 14.1 T for histidine sites, and B0 = 20.0 T for NAV. | |||
| His·HCl·H2O | NH3+ | 55.4 | 69.5 |
| Nδ | 90.2 | 94.9 | |
| Nε | 85.8 | 91.1 | |
| L-His | NH3+ | 58.4 | 59.7 |
| Nε | 105.4 | 105.9 | |
| NAV | N1 | 207.1 | 211.5 |
The δisoQ can be used to estimate the magnitude of the CQ and η at each site, using eqn (1)–(3). A contour plot of values of δisoQ at 14.1 T, calculated from eqn (1), as a function of the magnitude of the CQ and η are shown in Fig. S1 (ESI†). As expected from eqn (2), CQ and η are correlated, however at this field the δisoQ shows only a limited dependence on the asymmetry parameter η and thus CQ can be readily constrained to a range of ∼200 kHz irrespective of the asymmetry parameter. In L-His the NH3+ and Nε sites are determined to have CQ tensor magnitudes of 1.00–1.15 MHz and 1.37–1.58 MHz respectively, and the NH3+, Nε and Nδ sites in His·HCl·H2O have magnitudes of 1.00–1.15 MHz, 1.28–1.48 MHz and 1.32–1.52 MHz, for all possible values of η. The CQ of the NH3+ site in both compounds can be constrained to a smaller range of CQ magnitudes, since χq and, therefore, 14N δisoQ is less dependent on η when the CQ is smaller. The agreement of the 14N δisoQ determined from indirect detection methods in this paper, as compared to those calculated from literature values for 14N chemical shift and NQI tensors in the histidine compounds studied here is demonstrated in Table 2. In general, the 14N δisoQ observed in this work is smaller than that expected from literature values obtained by other techniques, or CASTEP calculated values. This discrepancy is most severe at the amine sites of both histidine molecules, with the His·HCl·H2O experimental NH3+ 14N δisoQ being 14.1 ppm lower than the calculated value, and that site in L-His was found to be 9 ppm lower than the literature/calculated value. At the remaining heterocyclic sites, the Nε site in His·HCl·H2O was found to deviate most from the 14N δisoQ value calculated from literature NQI parameter values, at 5.3 ppm. These deviations, being most severe for the highly mobile primary amine sites in histidine compounds, may be due to comparisons being made with NQI parameters determined by different techniques at cryogenic temperatures rather than room temperature, freezing out motions that could scale the 14N NQI in our experiments, or comparisons with CASTEP calculations that do not take into account molecular motions.
Quantitative analysis of histidine spectra
Having recorded indirect 14N detected spectra with very high efficiencies, the quality of data was high enough to observe subtle spectral features in the 14N lineshapes, for example, the shoulders on the sides of Nε and Nδ peaks in the His·HCl·H2O spectrum. As it is not possible to accurately determine CQ and η from the quadrupolar product alone, a more accurate analysis was undertaken fitting the 14N lineshapes in the indirect dimension. Numerical simulations were fitted to the experimentally determined lineshapes at each 14N site in the two histidine compounds, by systematically searching a CQ and η parameter space spanning CQ = ±0.25 MHz and η = ±0.25 of the published or calculated values. Plots of the root mean squared deviation (RMSD) between simulated and experimental spectra for each of the 14N sites in the histidine samples studied is shown in Fig. 3. The fits of these lineshapes were conducted with the quadrupolar and chemical shielding tensors oriented with respect to the 1H/14N dipolar coupling according the values given in Table 1. Simulations indicate that these parameters do indeed contribute to the overall 1H/14N lineshape, however for compounds with a small CQ these effects are negligible with the linewidths that are experimentally obtainable.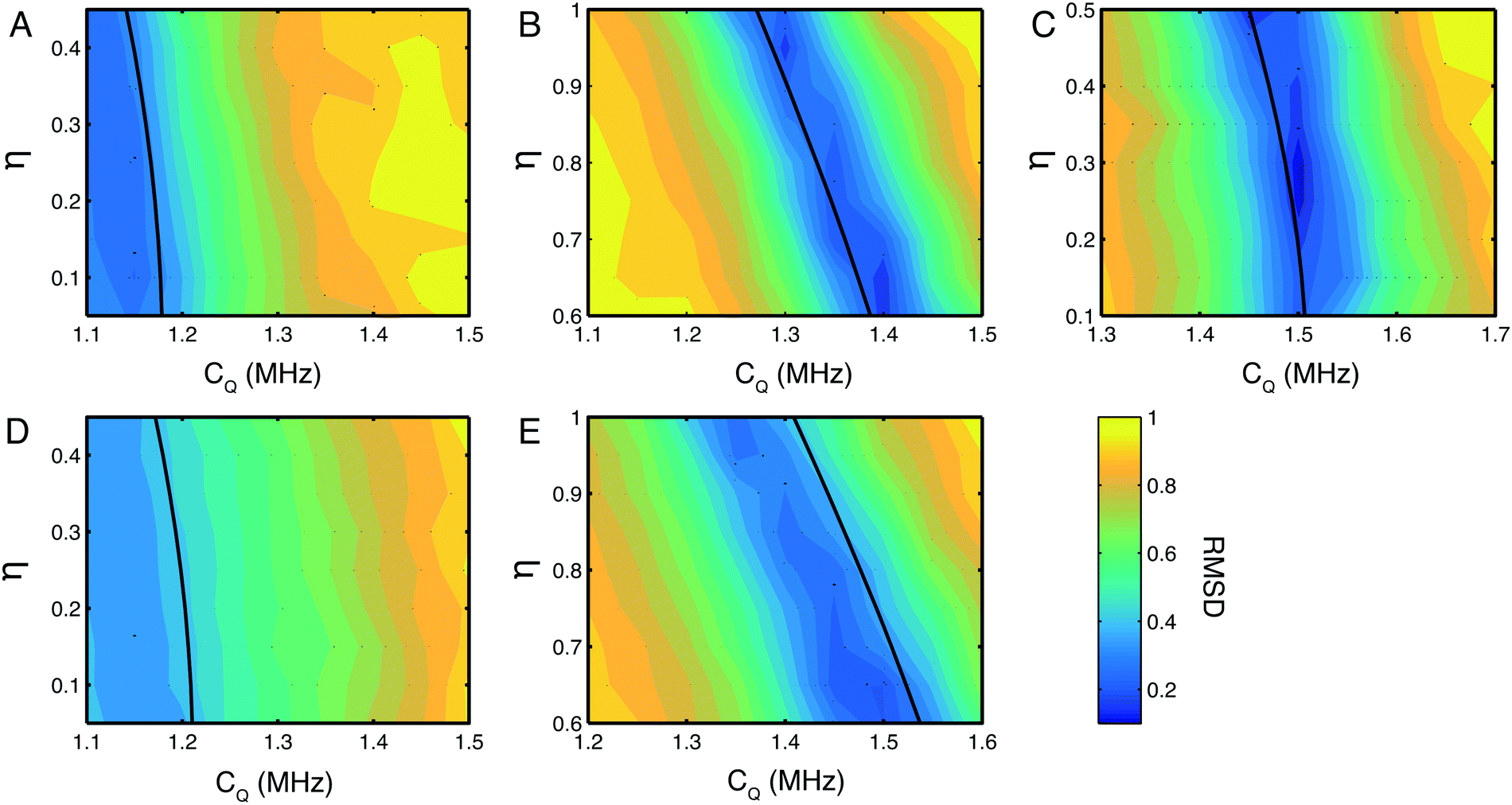 | ||
| Fig. 3 RMSD plots of simulated and experimental 14N lineshapes. Plots for the NH3+ (A) Nε (B) and Nδ (C) sites in His·HCl·H2O and the NH3+ (D) and Nε (E) sites in L-His. The magnitude of the CQ was arrayed in steps of 0.05 MHz from −0.25 to +0.25 MHz from the initial value, and η was arrayed in steps of 0.05 from −0.25 to +0.25 about a region spanning the predicted η for each nitrogen site based on the figures in Table 1. In all cases, contour lines are spaced by 5% of the maximum RMSD value. Errors estimated at the 10% RMSD level are CQ = 1.1 ± 0.3 MHz, η = 0.3 ± 0.3 for (A), CQ = 1.4 ± 0.08 MHz, η = 0.64 ± 0.04 for (B), CQ = 1.5 ± 0.1 MHz, η = 0.26 ± 0.06 for (C), CQ 1.1 ± 0.3 and η poorly defined (see text for discussion) for (D), CQ = 1.4 ± 0.15 MHz, η = 0.90 ± 0.06 for (E). The single black contour line in each plot indicates the δisoQ for each site calculated from the measured 14N shift δisoQ. | ||
There is a strip of relatively good agreement (RMSD < ∼0.3) through the RMSD plot for each 14N site where the simulated 14N δisoQ is consistent with that observed experimentally. A single black contour line through each plot marks the experimentally observed 14N δisoQ, which are also plotted on the contour plot in Fig. S1 (ESI†), and tabulated in Table 2. This line generally coincides with the area of lowest RMSD in each plot. This shows that the largest contribution to the quality of the simulations fit is determined from the 14N δisoQ, which is to be expected since it determines the position of the peak. In the RMSD plot for the Nδ of His·HCl·H2O, Fig. 3C, a single RMSD minimum where the RMSD is <0.15 (darkest blue contours) is observed. This covers a range of CQ = 1.49–1.51 MHz, and η = 0.22–0.34, which is in close agreement with the CASTEP calculated values at this site, CQ = 1.53 MHz, and η = 0.23, as well as literature values of CQ = 1.47 MHz, and η = 0.27.52 Additionally, visual comparison of the simulated lineshape with the experimental lineshape (Fig. 2A, yellow traces) shows a good agreement. This plot demonstrates that using a simulation fitting approach, one can define η to a range of ∼±0.1, which allows the CQ to be determined with a precision of tens of kHz, which is at least an order of magnitude more precise than one can achieve considering only the 14N δisoQ.
In the RMSD plots for the Nε sites of both His·HCl·H2O (Fig. 3B) and L-His (Fig. 3E), the fit of the lineshape with respect to η is somewhat less robust. In the case of the Nε site in His·HCl·H2O, two RMSD minima <0.2 are observed, at CQ = 1.29–1.3 MHz, η = 0.93–0.97, and CQ = 1.39–1.41 MHz, η = 0.60–0.68. The former of these minima, however, is consistent with the calculated values for this site, of CQ = 1.33 MHz, η = 0.94, as well as reported literature values of CQ = 1.29 MHz, η = 0.95.52 For the Nε site in L-His, the same general pattern is observed, with two RMSD minima in similar positions with respect to η as those observed at the same site in His·HCl·H2O (CQ = 1.35 MHz, η = 0.95–1.00, and CQ = 1.5 MHz, η = 0.60–0.65). However, the fit is not as good as that of the Nε site in the hydrochloride salt; in L-His the RMSD minima are broader, and the RMSD is not as low (minimum RMSD = 0.24). This is probably due to there being less well-defined features in the experimental lineshape at this site (as shown in Fig. 2B) compared to the Nε in His·HCl·H2O (Fig. 2A). However, the Nε sites are expected to have similar chemical environments and hydrogen bond geometries, and therefore similar 14N spectral parameters in terms of their 14N EFG and CSA tensors which give rise to the features observed in the 14N lineshapes, and one of the RMSD minima (at CQ = 1.35 MHz, η = 0.95–1.00) is consistent with expected values.
The NH3+ sites in both His·HCl·H2O and L-His show a poor fit of the simulated lineshape with respect to η, with the minimum RMSD encompassing almost the whole range of η values simulated, and not falling below RMSD = 0.3. This means it is not possible to use these plots to constrain the CQ at either site more precisely than was achieved by considering simply the 14N δisoQ.
In the case of the His·HCl·H2O the rather poorly defined minima reflects the featureless nature of the peak without any discontinuities, shoulders or splittings, making a distinctive fit difficult. This may be due to the fast 1H T2* decoherence at this site leading to a broadening of the signal and the loss of any distinctive features, indeed simulated spectra exhibited well defined splittings and shoulders before the application of the apodization to match the experimentally observed decay. We attribute this rapid relaxation to the molecular motions present at the site,53–55 which, in the case of NH3+ groups have previously been shown to influence the SQ 14N lineshape.18
At the NH3+ site in L-His, a well-defined splitting was observed in the 14N lineshape. However, the fit of the simulated spectra to this lineshape was the least satisfactory of all those investigated here, with a minimum RMSD = 0.35. As the experimental and simulated slices (Fig. 2B, red traces) show, the experimental spectrum observed was significantly broader, with more features, than the best fit simulated spectrum. The reason for this may be that at the NH3+, the 14N is coupled to three separate protons, which cannot be resolved in the 1H spectrum, and so their contributions to the 14N lineshape are superimposed in the 14N dimension. The simulated lineshape only takes into account an approximation of the spin system with 14N coupled to a single amine proton. This would mean that the simulation only accounted for part of the experimental lineshape, and that the remaining features were due to coupling to the other protons at the NH3+. Similar effects could also arise through the presence of conformational disorder in the sample or the presence of multiple polymorphs.
The differences observed between the fitted NH3+ lineshape and those experimentally obtained may also reflect the limitations of our current simulations which ignore the effects of relaxation and motional processes. Although such effects could be incorporated into the calculations, these simulations are already computationally demanding and, practically, close to the limit of what we can achieve in a reasonable timeframe (several weeks of supercomputer time) when fitting a grid of parameters to experimental data. In contrast; fits of the heterocyclic 14N sites exhibit a better fit with the experimental data, with the CQ and η in good agreement with published or calculated values. This is particularly clear for the Nδ site where the larger CQ is larger.
N-Acetyl-valine spectrum
Further proton detected experiments were performed on NAV (see Fig. 4), which exhibits a larger NQI than the sites studied in the histidine samples, with a CQ of 3.21 MHz and a η = 0.32,48 and as such mirrors the properties of 14N sites within the peptide bonds of proteins and peptides.56Fig. 4A shows a 2D 1H/14N correlation spectrum recorded on a sample of NAV at 20.0 T (850 MHz 1H Larmor frequency) and a MAS frequency of 78 kHz. Experiments were performed at a higher field with ultra-fast MAS in order to optimise conditions for proton detection on this challenging, high-CQ sample. The increased field and MAS frequency, significantly enhances the sensitivity of the experiment, lengthening coherence lifetimes and reducing the 1H homogeneous linewidth. The 14N shift of the amide 14N in NAV at this field is 335 ppm. From this shift, a 14N δisoQ = 207.3 ppm is calculated, by subtracting the δisocs of 127.7 ppm (measured by 15N CPMAS). This agrees well with the expected 14N δisoQ value of 211.5 ppm calculated for this field from literature values.48 The efficiency of the two-way 14N transfer is 9% with respect to the 1H spin echo signal under the same conditions. This is lower than the efficiencies of the proton detected experiment generally observed on the lower magnitude CQ sites in the histidine compounds. This is a trend observed in all indirect 14N detection experiments and can be attributed to the increased magnitude of the 14N CQ in NAV, which is likely to mean that the pulses applied on the 14N channel excite the far broader 14N spectrum to a lesser extent. | ||
| Fig. 4 1H/14N correlation spectrum of NAV at 20 T. Spectrum of NAV recorded at 78 kHz MAS. Experimental (red) and simulated (blue) projections through the 14N dimension are shown. (A) Plot of RMSD between experimental and simulated spectra of NAV at 20 T (B). | ||
Quantitative analysis of N-acetyl-valine spectra
A quantitative analysis of the NAV was conducted in a similar manner to that utilized for the histidine compounds. A plot of the 14N δisoQ, calculated from eqn (2), as a function of CQ and η at 20.0 T, with the experimentally measured NAV 14N δisoQ marked, is shown in Fig. S2 (ESI†). From this plot, one can read off the upper and lower limits placed on the CQ at the 14N site in NAV by the 14N δisoQ and determine the range of CQ magnitudes consistent with the 14N δisoQ as 2.83–3.26 MHz, for all values of η. This range of 430 kHz is far larger than the range of ∼200 kHz that could be obtained for the moderate CQ (1.0–1.5 MHz) sites found in histidines at 14.1 T. This is since when the CQ is large, the effects of η on the 14N δisoQ will be larger, as evident from eqn (2), since η and CQ are correlated, and the contribution of η is scaled by the CQ magnitude.Fig. 4B shows a contour plot of the RSMD of simulated spectra with respect to the experimental 14N lineshape in NAV as a function of the CQ and η used in the simulations. A comparison of the best-fit 14N simulated lineshape with the experimental lineshape is seen in the projections in Fig. 4. The quality of the fit is high, with a minimum RMSD = 1.5, however the RMSD minimum is relatively broad; covering a region of CQ magnitude of 75 kHz (CQ = 3.1 MHz–CQ = 3.175 MHz), and range of η values of 0.32 (0–0.32). The range of values covered by this RMSD minimum slightly underestimates the literature values of CQ = 3.21 MHz, η = 0.32.48 However, the black contour line which indicates the 14N δisoQ calculated from the measured 14N shift at the centre of gravity of the 14N lineshape is also underestimated by the best RMSD fit, and in fact agrees very well with the literature values. The lack of accuracy with which the simulations fit the data suggest that there are features of the experimental 14N lineshape that are not accounted for in the simulations. The reason for this is unclear. Simulations to ascertain if the relative orientations of the quadrupolar, dipolar and chemical shift anisotropy influenced the lineshape were conducted (see Fig. S3 and S4, ESI†). Simulations in the absence of the chemical shielding anisotropy (CSA) (Fig. S3, ESI†) show that the proton lineshape is strongly correlated to the β Euler angle describing the orientation of the quadrupolar tensor with respect to the 1H/14N dipolar coupling tensor, whilst the influence of the corresponding α Euler angle is limited to the lineshape. Similarly, introduction of the CSA into the simulations (Fig. S4, ESI†) resulted in minor changes to the lineshape. Although the effects of tensor orientation are pronounced when the linewidths are small, many of these factors are masked with the currently attainable experimental resolution, which together with the computationally demanding nature of the simulations and large parameter space precludes a more rigorous statistical fitting of the lineshape. Alternatively, it is plausible that the signal is broadened or shifted by higher-order terms (>2) in the quadrupolar Hamiltonian that are not accounted for in the simulations and not removed by MAS. For sites with lower (<1.5 MHz) quadrupolar couplings, it was verified that these terms did not affect the simulated lineshapes. However, this was not verified for the case where the quadrupolar coupling was large (∼3 MHz) and high order terms are expected to be more significant, as running the simulations with the full Hamiltonian was found to be prohibitively time consuming. Finally, it may be that the 14N spectrum of this site is broadened by high mobility of the amide nitrogen, although we acknowledge that this is not reflected in the size of the quadrupolar interaction.
Multidimensional 14N correlation spectroscopy
The quadrupolar interaction provides a sensitive reporter of secondary structure within proteins, with variations of up to 200 kHz reported between alpha-helical and beta-sheet structures, corresponding to variation in δisoQ of ∼130 ppm in the proton detected spectra reported here. Despite the large variation in δisoQ reported for proteins, for larger biomolecules its application will remain challenging due to the limited chemical shift dispersion in the amide protons. Although further enhancements in resolution and sensitivity can be realised through the application of faster MAS and higher 14N rf fields,26,32 for larger biomolecules it is clear that higher-dimensionality spectra will be required to aid resolution and assignment. In Fig. 1B we describe a 3D experiments which allows the acquisition of a 14N resolved 1H/1H correlation experiment. Analogous in many ways to the 15N filtered 1H/1H NOESY experiment frequently employed in the liquid state, the 1H/1H correlations are now resolved according to the δisoQ of the adjacent 14N site. The efficiency of the experiment ensures that such data sets can be acquired in as little as 2 days, with the overall duration limited by the phase cycling employed (Fig. 5).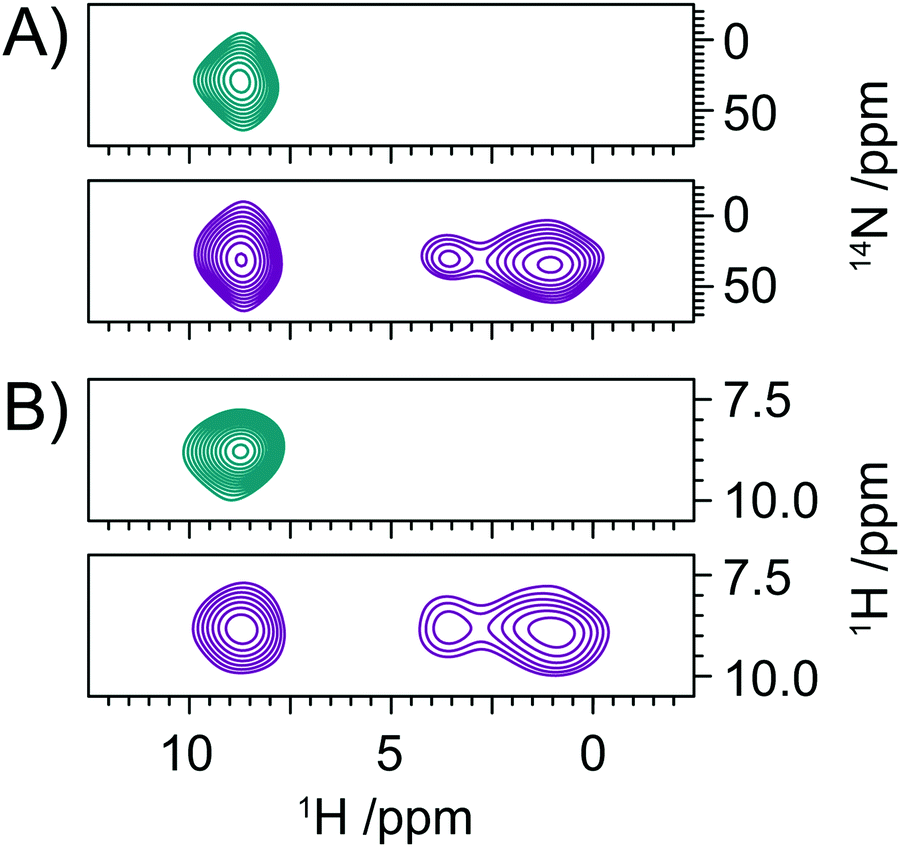 | ||
| Fig. 5 Slices extracted from a 3D, 14N resolved 1H/1H correlation spectrum of valine with no proton/proton mixing (teal), and 24 rotor cycles of RFDR mixing (purple). 1H/14N planes (A) and their respective 1H/1H planes (B). | ||
Conclusions
We have demonstrated the application of a novel method of indirect detection of 14N via1H under moderate and fast MAS frequencies, applied to two forms of histidine and NAV. Spectra with up to 27.5% efficiency can be recorded using this method, resulting in a large increase in S/N and spectral quality, over previous methods. The data presented have been fitted to simulated lineshapes to extract NQI parameters for nitrogen sites in different chemical environments with increased precision. The 1H detected 14N experiments described here are not as limited in the number of sites that can studied as previous methods such as piecewise detection, indeed the variation observed in CQ served to enhance the resolution. Furthermore, exploitation of 1H detection facilitates its application to unlabelled biomolecules, providing a viable route to study the structure and dynamics complex natural organic and biomolecular systems.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by The Wellcome Trust (PTFW and MC), EPSRC (EP/H003789/1, IK; EP/M023664/1, MC/PTFW/IK), The University of Southampton (JJ), and The Royal Society University Research Fellowship (MC). IK thanks EPSRC for an Impact Acceleration grant, and Jos Martin of MathWorks for technical support. IH thanks The Higher Committee for Education Development in Iraq for financial support. The UK 850 MHz solid-state NMR Facility instruments used in this research were funded by EPSRC and BBSRC, as well as the University of Warwick, including via part funding through Birmingham Science City Advanced Materials Projects 1 and 2 supported by Advantage West Midlands (AWM) and the European Regional Development Fund (ERDF). The authors acknowledge the use of the IRIDIS High Performance Computing Facility, and associated support services at the University of Southampton, in the completion of this work.References
- K. J. Harris, S. L. Veinberg, C. R. Mireault, A. Lupulescu, L. Frydman and R. W. Schurko, Chemistry, 2013, 19, 16469–16475 CrossRef CAS PubMed.
- L. A. O'dell, R. W. Schurko, K. J. Harris, J. Autschbach and C. I. Ratcliffe, J. Am. Chem. Soc., 2011, 133, 527–546 CrossRef PubMed.
- S. L. Veinberg, Z. W. Friedl, K. J. Harris, L. A. O'Dell and R. W. Schurko, CrystEngComm, 2015, 17, 5225–5236 RSC.
- R. Tycko and S. J. Opella, J. Am. Chem. Soc., 1986, 108, 3531–3532 CrossRef CAS.
- R. Tycko and S. J. Opella, J. Chem. Phys., 1987, 86, 1761 CrossRef CAS.
- R. Tycko, P. L. Stewart and S. J. Opella, J. Am. Chem. Soc., 1986, 108, 5419–5425 CrossRef CAS.
- L. A. O'Dell and A. Brinkmann, J. Chem. Phys., 2013, 138, 064201 CrossRef PubMed.
- L. A. O'Dell, R. He and J. Pandohee, CrystEngComm, 2013, 15, 8657 RSC.
- L. A. O'dell and C. I. Ratcliffe, Chem. Phys. Lett., 2011, 514, 168–173 CrossRef.
- A. J. Rossini, L. Emsley and L. A. O'Dell, Phys. Chem. Chem. Phys., 2014, 16, 12890–12899 RSC.
- I. M. Haies, J. A. Jarvis, H. Bentley, I. Heinmaa, I. Kuprov, P. T. F. Williamson and M. Carravetta, Phys. Chem. Chem. Phys., 2015, 17, 6577–6587 RSC.
- Y. Nishiyama, M. Malon, Z. Gan, Y. Endo and T. Nemoto, J. Magn. Reson., 2013, 230, 160–164 CrossRef CAS PubMed.
- I. Haies, J. Jarvis, L. J. Brown, I. Kuprov, P. T. F. Williamson and M. Carravetta, Phys. Chem. Chem. Phys., 2015, 17, 23748–23753 RSC.
- Z. H. Gan, J. Am. Chem. Soc., 2006, 128, 6040–6041 CrossRef CAS PubMed.
- Z. H. Gan, J. Magn. Reson., 2007, 184, 39–43 CrossRef CAS PubMed.
- Z. H. Gan, J. P. Amoureux and J. Trebosc, Chem. Phys. Lett., 2007, 435, 163–169 CrossRef CAS.
- S. Cavadini, A. Abraham and G. Bodenhausen, J. Magn. Reson., 2008, 190, 160–164 CrossRef CAS PubMed.
- S. Cavadini, A. Abraham, S. Ulzega and G. Bodenhausen, J. Am. Chem. Soc., 2008, 130, 10850–10851 CrossRef CAS PubMed.
- S. Cavadini, S. Antonijevic, A. Lupulescu and G. Bodenhausen, ChemPhysChem, 2007, 8, 1363–1374 CrossRef CAS PubMed.
- S. Cavadini, S. Antonijevic, A. Lupulescu and G. Bodenhausen, J. Magn. Reson., 2006, 182, 168–172 CrossRef CAS PubMed.
- S. Cavadini, A. Lupulescu, S. Antonijevic and G. Bodenhausen, J. Am. Chem. Soc., 2006, 128, 7706–7707 CrossRef CAS PubMed.
- S. Cavadini, V. Vitzthum, S. Ulzega, A. Abraham and G. Bodenhausen, J. Magn. Reson., 2010, 202, 57–63 CrossRef CAS PubMed.
- V. Vitzthum, M. A. Caporini and G. Bodenhausen, J. Magn. Reson., 2010, 205, 177–179 CrossRef CAS PubMed.
- V. Vitzthum, M. A. Caporini, S. Ulzega and G. Bodenhausen, J. Magn. Reson., 2011, 212, 234–239 CrossRef CAS PubMed.
- V. Vitzthum, M. A. Caporini, S. Ulzega, J. Trebosc, O. Lafon, J. P. Amoureux and G. Bodenhausen, J. Magn. Reson., 2012, 223, 228–236 CrossRef CAS PubMed.
- Y. L. Hong, T. Asakura and Y. Nishiyama, ChemPhysChem, 2018, 19, 1841–1845 CrossRef CAS PubMed.
- G. N. M. Reddy, M. Malon, A. Marsh, Y. Nishiyama and S. P. Brown, Anal. Chem., 2016, 88, 11412–11419 CrossRef CAS PubMed.
- D. Carnevale, X. Ji and G. Bodenhausen, J. Chem. Phys., 2017, 147, 184201 CrossRef PubMed.
- A. S. Tatton, T. N. Pham, F. G. Vogt, D. Iuga, A. J. Edwards and S. P. Brown, Mol. Pharmaceutics, 2013, 10, 999–1007 CrossRef CAS PubMed.
- A. S. Tatton, T. N. Pham, F. G. Vogt, D. Iuga, A. J. Edwards and S. P. Brown, CrystEngComm, 2012, 14, 2654 RSC.
- Y. Nishiyama, Y. Endo, T. Nemoto, H. Utsumi, K. Yamauchi, K. Hioka and T. Asakura, J. Magn. Reson., 2011, 208, 44–48 CrossRef CAS PubMed.
- J. A. Jarvis, I. M. Haies, P. T. F. Williamson and M. Carravetta, Phys. Chem. Chem. Phys., 2013, 15, 7613–7620 RSC.
- D. Marion, M. Ikura, R. Tschudin and A. Bax, J. Magn. Reson., 1989, 85, 393–399 CAS.
- A. E. Bennett, J. H. Ok, R. G. Griffin and S. Vega, J. Chem. Phys., 1992, 96, 8624–8627 CrossRef CAS.
- F. Delaglio, S. Grzesiek, G. W. Vuister, G. Zhu, J. Pfeifer and A. Bax, J. Biomol. NMR, 1995, 6, 277–293 CrossRef CAS PubMed.
- W. F. Vranken, W. Boucher, T. J. Stevens, R. H. Fogh, A. Pajon, P. Llinas, E. L. Ulrich, J. L. Markley, J. Ionides and E. D. Laue, Proteins, 2005, 59, 687–696 CrossRef CAS PubMed.
- P. Bertani, J. Raya and B. Bechinger, Solid State Nucl. Magn. Reson., 2014, 61–62, 15–18 CrossRef CAS PubMed.
- H. J. Hogben, M. Krzystyniak, G. T. P. Charnock, P. J. Hore and I. Kuprov, J. Magn. Reson., 2011, 208, 179–194 CrossRef CAS PubMed.
- M. Profeta, F. Mauri and C. J. Pickard, J. Am. Chem. Soc., 2003, 125, 541–548 CrossRef CAS PubMed.
- S. J. Clark, M. D. Segall, C. J. Pickard, P. J. Hasnip, M. J. Probert, K. Refson and M. C. Payne, Z. Kristallogr., 2005, 220, 567–570 CAS.
- C. J. Pickard and F. Mauri, Phys. Rev. B: Condens. Matter Mater. Phys., 2001, 63, 245101 CrossRef.
- J. R. Yates, C. J. Pickard and F. Mauri, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 76, 024401 CrossRef.
- S. Li, L. Zhou, Y. Su, B. Han and F. Deng, Solid State Nucl. Magn. Reson., 2013, 54, 13–17 CrossRef CAS PubMed.
- S. Li and M. Hong, J. Am. Chem. Soc., 2011, 133, 1534–1544 CrossRef CAS PubMed.
- D. K. Lee, Y. F. Wei and A. Ramamoorthy, J. Phys. Chem. B, 2001, 105, 4752–4762 CrossRef CAS.
- Y. F. Wei, D. K. Lee, A. E. McDermott and A. Ramamoorthy, J. Magn. Reson., 2002, 158, 23–35 CrossRef CAS PubMed.
- J.-S. Lee and A. K. Khitrin, Concepts Magn. Reson., Part A, 2008, 32, 56–67 CrossRef.
- R. E. Stark, R. A. Haberkorn and R. G. Griffin, J. Chem. Phys., 1978, 68, 1996 CrossRef CAS.
- M. D. Esrafili, H. Behzadi and N. L. Hadipour, Biophys. Chem., 2008, 133, 11–18 CrossRef CAS PubMed.
- M. Bak, R. Schultz, T. Vosegaard and N. C. Nielsen, J. Magn. Reson., 2002, 154, 28–45 CrossRef CAS PubMed.
- M. K. Pandey and Y. Nishiyama, J. Magn. Reson., 2015, 258, 96–101 CrossRef CAS PubMed.
- C. A. Mcdowell, A. Naito, D. L. Sastry and K. Takegoshi, J. Magn. Reson., 1986, 69, 283–292 CAS.
- S. E. Ashbrook, S. Antonijevic, A. J. Berry and S. Wimperis, Chem. Phys. Lett., 2002, 364, 634–642 CrossRef CAS.
- M. J. Thrippleton, M. Cutajar and S. Wimperis, Chem. Phys. Lett., 2008, 452, 233–238 CrossRef CAS.
- M. Chan-Huot, S. Wimperis, C. Gervais, G. Bodenhausen and L. Duma, ChemPhysChem, 2015, 16, 204–215 CrossRef CAS PubMed.
- J. A. Jarvis, I. Haies, M. Lelli, A. J. Rossini, I. Kuprov, M. Carravetta and P. T. F. Williamson, Chem. Commun., 2017, 53, 12116–12119 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8cp06276e |
| ‡ Current address: Department of Life Sciences, Imperial College, SW7 2AZ, London, UK. |
| This journal is © the Owner Societies 2019 |