
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDiscriminating Seebeck sensing of molecules†
Hatef
Sadeghi
 *
*
Physics Department, Lancaster University, Lancaster LA1 4YB, UK. E-mail: h.sadeghi@lancaster.ac.uk
First published on 7th January 2019
Abstract
One of the fundamental challenges in molecular-scale sensors is the junction to junction variability leading to variations in their electrical conductance by up to a few orders of magnitude. In contrast, thermal voltage measurements of single and many molecule junctions show that this variation in the Seebeck coefficient is smaller. Particularly, the sign of the Seebeck coefficient is often resilient against conformational changes. In this paper, we demonstrate that this robust molecular feature can be utilised in an entirely new direction of discriminating molecular sensing of gas and bio-molecules. We show that the positive sign of the Seebeck coefficient in the presence of cytosine nucleobases changes to a negative one when cancerous cytosine nucleobases were absorbed on the molecular wire formed by metalloporphyrins. Furthermore, the sign of the Seebeck coefficient changes when chlorine gas interacts with the Mn-porphyrin molecular wire. The change in the sign of Seebeck coefficient is due to the formation of spin driven bound states with energies close to the Fermi energy of electrodes. Seebeck sensing is a generic concept and opens new avenues for molecular sensing with huge potential applications in the years ahead.
Introduction
The search for molecular sensors based on changes in their electrical or ionic currents have been subject to many studies in the last couple of decades.1 However, a weak signal-to-noise ratio and a large junction to junction variation have limited their application. From a fundamental science perspective, many interesting properties such as quantum interference,2 photoswitching3 and electronic switching4 have been observed in single molecules using mechanically controllable (MCBJ) and scanning tunneling microscopy break-junction (STMBJ) methods. In these methods, many molecular junctions are formed and the logarithm of the obtained conductance histogram is fitted to a Lorentzian function. The mean value is reported as the statistically most probable molecular conductance.5 The conductance variation (the width of the fitted Lorentzian function) can be as high as a couple of orders of magnitude.5 This variation is mainly due to the conformational changes in molecules, their binding configurations to electrodes and the effect of the surrounding environment.6,7 In any electrical signal based molecular sensing device, changes in the electrical conductance upon interaction with the target molecule “analyte” are used for molecular recognition. Molecular junctions usually pose low conductances. Therefore their sensitivity can be largely affected by the unwanted variations in their electrical conductance which in turn can mask the analyte signature.Results and discussion
In this paper, we demonstrate that molecular-scale thermoelectricity can be utilised for selective molecular sensing. This includes discriminating sensing of molecules such as DNA bases or gases using changes in the sign of Seebeck coefficient (Seebeck sensing) upon their interactions with a molecular backbone in a junction. Using the Seebeck coefficient for sensing is advantageous compared with methods based on electrical sensing, because the electrical conductance (G) depends on the magnitude of the transmission probability T(E), of electrons with energy E passing from one electrode to the other through a molecular backbone (G ∝ T(EF)), whereas from the Mott formula8 the Seebeck coefficient is proportional to the slope of T(E) at Fermi energy EF where kB, T and e are the Boltzmann constant, temperature and electron charge, respectively. Therefore, two analytes that possess similar conductances can have Seebeck coefficients of different signs or amplitude. Furthermore, the electrical conductances of molecules such as nucleobases or gases are extremely low which is problematic for conductance-based sensing, but advantageous for Seebeck sensing, since a low conductance typically leads to a high Seebeck coefficient. From the Mott formula, if the Fermi energy is close to the energy of the highest occupied molecular orbitals (HOMO dominated molecules), S is positive whereas in LUMO (lowest unoccupied molecular orbital) dominated molecules, it is negative (Fig. 1). If the transport mechanism changes from a HOMO dominated transport in the absence of analyte to a LUMO dominated one in the presence of analyte, regardless of the magnitude of the changes in G or S, the analyte can be recognised.
the Seebeck coefficient is proportional to the slope of T(E) at Fermi energy EF where kB, T and e are the Boltzmann constant, temperature and electron charge, respectively. Therefore, two analytes that possess similar conductances can have Seebeck coefficients of different signs or amplitude. Furthermore, the electrical conductances of molecules such as nucleobases or gases are extremely low which is problematic for conductance-based sensing, but advantageous for Seebeck sensing, since a low conductance typically leads to a high Seebeck coefficient. From the Mott formula, if the Fermi energy is close to the energy of the highest occupied molecular orbitals (HOMO dominated molecules), S is positive whereas in LUMO (lowest unoccupied molecular orbital) dominated molecules, it is negative (Fig. 1). If the transport mechanism changes from a HOMO dominated transport in the absence of analyte to a LUMO dominated one in the presence of analyte, regardless of the magnitude of the changes in G or S, the analyte can be recognised.
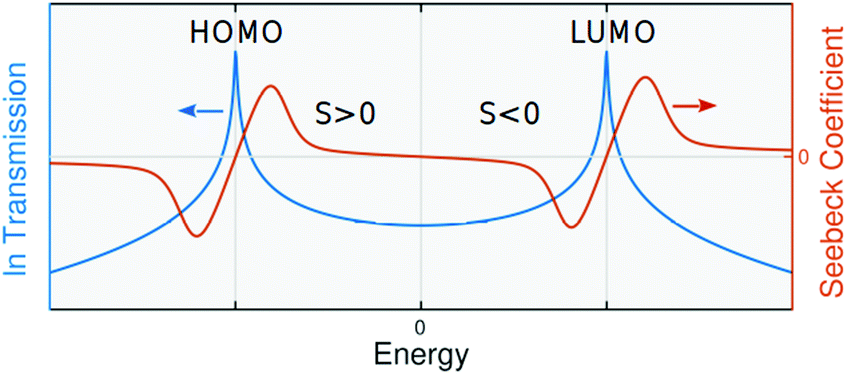 | ||
| Fig. 1 Seebeck and transmission coefficients. Seebeck coefficient is proportional to the slope of transmission coefficient. In HOMO dominated molecules, the sign of Seebeck coefficient is positive whereas the sign of Seebeck coefficient is negative in LUMO dominated molecules. | ||
Unlike the amplitude of electrical conductance and Seebeck coefficient, the sign of Seebeck coefficient is robust and less affected by molecular conformation. For example several research groups9,10 have reported a positive Seebeck coefficient for molecular junctions formed by 1,4-benzenedithiol (BDT) sandwiched between two gold electrodes (Au/BDT/Au) using different experimental techniques such as STMBJ,11–14 MCBJ15,16 and conducting probe atomic force microscopy.17,18 A positive Seebeck coefficient has also been found in Au/dibenzenedithiol/Au,12,19 Au/tribenzenedithiol/Au,12,19 Au/1,4-n-phenylenediamine/Au18 and Au/thiophene/Au20 molecular junctions. On the other hand, a negative Seebeck coefficient has been observed in Au/C60/Au,11,21–23 Au/4,4-bipyridine/Au24,25 and Au/BPE/Au.24,25 The sign of Seebeck coefficient does not even change in Au/biphenyl-4,4′-dithiol/Au and Au/C60/Au junctions upon applying a gate voltage in the range of ±8 V.23 Although these experiments have been carried out using different techniques and samples, they all measure the same sign of S. It is crucial that not only the most probable thermal voltage but also the distribution of S including less probable conformations show the same sign as that of the peak value (see Fig. S1 of the ESI†). In what follows, our aim is to demonstrate that this robust feature can be utilized for molecular sensing.
We form a junction using metalloporphyrin molecules with a manganese Mn-II (MnP) metallic center connected to graphene electrodes through pyrene anchors (Fig. 2a). Due to partially filled d orbitals of manganese, the occupancy of majority and minority spins is different and the junction is spin polarized. Using the mean field Hamiltonian obtained from density functional theory (DFT), we calculate the spin dependent transmission coefficient Tσ(E) of the MnP junction where σ = [↑, ↓] (see computational methods). Fig. 2b shows the total transmission coefficient T(E) = (T↓(E) + T↑(E))/2 (see Fig. S2 of the ESI† for T↓(E) and T↑(E)). Clearly the LUMO resonance is closer to the DFT Fermi energy (E = 0 eV). Therefore, the Seebeck coefficient is negative in the vicinity of the DFT Fermi energy as shown in Fig. 2c. In the presence of chlorine (Cl) atom (which is a gas at room temperature), the Cl atom is bonded to manganese (Mn) to form Mn-III (MnP–Cl).26 Consequently, transmission through the MnP–Cl junction is dominated by HOMO (Fig. 2b) leading to a positive Seebeck coefficient around the DFT Fermi energy (Fig. 2c). In contrast, in the presence of a nitrogen molecule (N2), the sign of Seebeck coefficient does not change (Fig. S3 of the ESI†). Clearly, the sign of Seebeck coefficient alone can be used to discriminate chlorine from nitrogen gas. It is worth mentioning that the Seebeck coefficient is enhanced significantly in the MnP–Cl junction to ∼400 μV K−1 compared to ∼150 μV K−1 in the MnP junction at DFT Fermi energy. Particularly while the sign of S is sensitive to chlorine in the MnP junction, it does not change if a zinc-porphyrin junction is used as the sensing backbone (see Fig. S5 of the ESI†). Therefore, a suitable choice of backbone should be made for sensing of a given analyte.
 | ||
| Fig. 2 Mn-porphyrin molecular junction. (a) Mn-porphyrin between the hot and cold graphene electrodes in the presence of chlorine. (b) Transmission coefficient T(E) and (c) Seebeck coefficient S in the absence and presence of chlorine. Total transmission coefficient is a sum of transmission due to the majority and minority spins (see Fig. S2 of the ESI† for spin dependent transmission functions). The transmission is LUMO dominated for the bare junction. In the presence of chlorine, the transmission is through HOMO, therefore the sign of Seebeck coefficient changes. (d) The spin density of MnP–Cl with pyrene anchors where the majority spins are localised on MnCl. (e) A model calculation where a backbone with two sites is connected to two 1D leads. An analyte is modelled by a site energy bonded to the backbone which creates a resonance in the HOMO–LUMO gap of the backbone. (f) Transmission coefficient and (g) Seebeck coefficient in the absence (blue cure) and presence (red curve) of an analyte. | ||
The spin density calculation shows that the majority spins are localised on MnCl (Fig. 2d) forming a new HOMO close to the Fermi energy (Fig. 2b). When a bound state (e.g. Cl states) interacts with the continuum of states (e.g. MnP), due to the interference between transmitted and reflected waves, a Fano resonance is formed8 (see Fig. S4 of the ESI†). This is demonstrated using a simple model where a scatterer with two sites is attached to two one dimensional leads (Fig. 2e). Fig. 2f shows the corresponding transmission coefficient. In the absence of the analyte, HOMO and LUMO resonances are formed (blue curve in Fig. 2f). The transport is LUMO dominated since the LUMO resonance is closer to the electrode's Fermi energy (E = 0 eV). In contrast, in the presence of the analyte (red curve in Fig. 2f), a Fano resonance and consequently a new HOMO is formed. As shown in Fig. 2g, the negative sign of Seebeck coefficient (LUMO dominated transport) changes to a positive one in the presence of the analyte (HOMO dominated transport).
In order to understand why the sign of Seebeck coefficient can change, consider a one level system (e.g. a frontier molecular orbital) connected to two one dimensional leads (Fig. S4a of ESI†). The transmission coefficient of this one level system is obtained using the Breit–Wigner formula:8
 | (1) |
 | (2) |
The sign and amplitude of the Seebeck coefficient can also be used for selective sensing of DNA bases. The molecular junction formed by Ni-porphyrin molecules attached to graphene electrodes through π–π stacking to the hexabenzocoronene HBC anchors (Fig. 3a) shows a positive Seebeck coefficient around the DFT Fermi energy (Fig. 3c). The sign of S does not change when cytosine nucleobases physisorbed on porphyrin (Fig. 3c) because the Fano-resonance due to the cytosine happens to be at ∼0.5 eV away from the Fermi energy (Fig. 3b). In addition, a narrow Fano-resonance indicates a weak coupling between the cytosine and the porphyrin backbone. In contrast, if a cancerous nucleobase, cyanocytosine, is physisorbed on porphyrin, the sign of S changes and the amplitude of S increases significantly up to an order of magnitude. This is because the Fano-resonance due to the cancerous nucleobase happens to be close to the Fermi energy. The width of the Fano-resonance is also larger due to the stronger coupling between the cancerous nucleobases and the porphyrin backbone. It is interesting to note that the sign of S at the DFT Fermi energy is not affected by temperature in NiP junctions for a wide temperature range between 100 K and 400 K (Fig. 3d). Clearly, the sign of the Seebeck coefficient alone can be utilized to distinguish cancerous cytosine nucleobases from the healthy cytosine nucleobases.
 | ||
| Fig. 3 Ni-porphyrin molecular junction. (a) Ni-porphyrin between the graphene electrodes in the absence and presence of the healthy cytosine DNA nucleotide and cancerous cyanocytosine.27,28 (b) Transmission coefficient T. (c) Seebeck coefficient S in the absence and presence of cytosine and cyanocytosine versus Fermi energy EF at room temperature. (d) S versus temperature at the DFT Fermi energy EF = 0 eV. The transmission is LUMO dominated for the bare junction. In the presence of cytosine, the transmission become HOMO dominated, therefore the sign of Seebeck coefficient changes. In contrast, in the presence of cyanocytosine, the transmission remains LUMO dominated with a negative Seebeck coefficient. | ||
Conclusions
We demonstrated that the sign of Seebeck coefficient which is often robust against conformational changes can be employed for discriminating sensing of gas molecules or biological species. Discriminating sensing of molecules using changes in the sign of Seebeck coefficient opens encouraging perspectives for the realization of real time, fast and portable devices for the sensing of gas and biomolecules.Computational methods
Geometry optimization
The geometry of each structure was relaxed to the force tolerance of 10 meV Å−1 using the SIESTA29 implementation of density functional theory (DFT), with a double-ζ polarized basis set (DZP) and the generalized gradient approximation (GGA) functional with Perdew–Burke–Ernzerhof (PBE) parameterization. A real-space grid was defined with an equivalent energy cut-off of 250 Ry.Electrons transport
To calculate electronic properties of the device, from the converged DFT calculation, the underlying mean-field Hamiltonian H was combined with our quantum transport code, Gollum.30 This yields the transmission coefficient Tel(E) for electrons of energy E (passing from the source to the drain) via the relation T(E) = Tr(ΓL(E)GR(E)ΓR(E)GR†(E)) where describes the level broadening due to the coupling between the left L and the right R electrodes and the central scattering region, ΓL,R(E) are the retarded self-energies associated with this coupling and
describes the level broadening due to the coupling between the left L and the right R electrodes and the central scattering region, ΓL,R(E) are the retarded self-energies associated with this coupling and 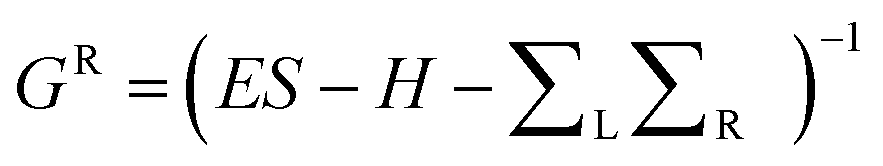 is the retarded Green's function, where H is the Hamiltonian and S is the overlap matrix obtained from SIESTA implementation of DFT. The DFT + ∑ approach has been employed for spectral adjustment.8 In the case of spin-polarised calculations, the total transmission is obtained from Ttot = (T↑ + T↓)/2 where T↑ and T↓ are spin up and down transmissions.
is the retarded Green's function, where H is the Hamiltonian and S is the overlap matrix obtained from SIESTA implementation of DFT. The DFT + ∑ approach has been employed for spectral adjustment.8 In the case of spin-polarised calculations, the total transmission is obtained from Ttot = (T↑ + T↓)/2 where T↑ and T↓ are spin up and down transmissions.
Thermoelectric properties
The electrical conductance G(T) = G0L0, and the Seebeck coefficient S(T) = −L1/eTL0 are calculated from the electron transmission coefficient T(E) where the momentums, and f(E,T) is the Fermi–Dirac probability distribution function, f(E,T) = (e(E−EF)/kBT + 1)−1, where T is the temperature, kB is the Boltzmann constant, EF is the Fermi energy, G0 = 2e2/h is the conductance quantum, e is electron charge and h is the Planck's constant.
and f(E,T) is the Fermi–Dirac probability distribution function, f(E,T) = (e(E−EF)/kBT + 1)−1, where T is the temperature, kB is the Boltzmann constant, EF is the Fermi energy, G0 = 2e2/h is the conductance quantum, e is electron charge and h is the Planck's constant.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
H. S. acknowledges the Leverhulme Trust for Early Career Fellowship no. ECF-2017-186.Notes and references
- J. J. Gooding and K. Gaus, Angew. Chem., Int. Ed., 2016, 55, 11354–11366 CrossRef CAS PubMed.
- S. Sangtarash, C. Huang, H. Sadeghi, G. Sorohhov, J. Hauser, T. Wandlowski, W. Hong, S. Decurtins, S. X. Liu and C. J. Lambert, J. Am. Chem. Soc., 2015, 137, 11425–11431 CrossRef CAS PubMed.
- M. Galperin and A. Nitzan, Phys. Chem. Chem. Phys., 2012, 14, 9421 RSC.
- E. Lörtscher, J. W. Ciszek, J. Tour and H. Riel, Small, 2006, 2, 973–977 CrossRef PubMed.
- F. Schwarz and E. Lörtscher, J. Phys.: Condens. Matter, 2014, 26, 474201 CrossRef PubMed.
- S. V. Aradhya and L. Venkataraman, Nat. Nanotechnol., 2013, 8, 399–410 CrossRef CAS PubMed.
- Q. H. Al-Galiby, H. Sadeghi, D. Z. Manrique and C. J. Lambert, Nanoscale, 2017, 9, 4819–4825 RSC.
- H. Sadeghi, Nanotechnology, 2018, 29, 373001 CrossRef PubMed.
- L. Cui, R. Miao, C. Jiang, E. Meyhofer and P. Reddy, J. Chem. Phys., 2017, 146, 092201 CrossRef.
- L. Rincón-García, C. Evangeli, G. Rubio-Bollinger and N. Agraït, Chem. Soc. Rev., 2016, 45, 4285–4306 RSC.
- S. K. Lee, T. Ohto, R. Yamada and H. Tada, Nano Lett., 2014, 14, 5276 CrossRef CAS PubMed.
- J. A. Malen, P. Doak, K. Baheti, T. D. Tilley, A. Majumdar and R. A. Segalman, Nano Lett., 2009, 9, 3406–3412 CrossRef CAS PubMed.
- Y. Kim, A. Lenert, E. Meyhofer and P. Reddy, Appl. Phys. Lett., 2016, 109, 10–14 Search PubMed.
- P. Reddy, S. Y. Jang, R. A. Segalman and A. Majumdar, Science, 2007, 315, 1568–1571 CrossRef CAS PubMed.
- M. Tsutsui, M. Taniguchi and T. Kawai, Nano Lett., 2008, 8, 3293–3297 CrossRef CAS PubMed.
- T. Morikawa, A. Arima, M. Tsutsui and M. Taniguchi, Nanoscale, 2014, 6, 8235–8241 RSC.
- A. Tan, S. Sadat and P. Reddy, Appl. Phys. Lett., 2010, 96, 013110 CrossRef.
- S. Y. Jang, P. Reddy, A. Tan, J. Balachandran, S. Sadat, V. Gavini, B. D. Dunietz, S. Y. Jang and P. Reddy, J. Am. Chem. Soc., 2011, 133, 8838–8841 CrossRef PubMed.
- B. Xu and N. J. Tao, Science, 2003, 301, 1221–1223 CrossRef CAS PubMed.
- W. B. Chang, C. K. Mai, M. Kotiuga, B. Neaton, G. C. Bazan, R. A. Segalman, J. B. Neaton, G. C. Bazan and R. A. Segalman, Chem. Mater., 2014, 26, 7229–7235 CrossRef CAS.
- S. K. Yee, J. A. Malen, A. Majumdar and R. A. Segalman, Nano Lett., 2011, 11, 4089–4094 CrossRef CAS PubMed.
- C. Evangeli, K. Gillemot, E. Leary, M. T. González, G. Rubio-Bollinger, C. J. Lambert and N. Agraït, Nano Lett., 2013, 13, 2141–2145 CrossRef CAS PubMed.
- Y. Kim, W. Jeong, K. Kim, W. Lee and P. Reddy, Nat. Nanotechnol., 2014, 9, 881–885 CrossRef CAS PubMed.
- T. Kim, P. Darancet, J. R. Widawsky, M. Kotiuga, S. Y. Quek, J. B. Neaton and L. Venkataraman, Nano Lett., 2014, 14, 794–798 CrossRef CAS PubMed.
- J. R. Widawsky, P. Darancet, J. B. Neaton and L. Venkataraman, Nano Lett., 2012, 12, 354–358 CrossRef CAS PubMed.
- E. Rose, B. Andrioletti, S. Zrig and M. Quelquejeu-Ethève, Chem. Soc. Rev., 2005, 34, 573–583 RSC.
- T. Ahmed, J. T. Haraldsen, J. X. Zhu and A. V. Balatsky, J. Phys. Chem. Lett., 2014, 5, 2601–2607 CrossRef CAS PubMed.
- R. Chahwan, S. N. Wontakal and S. Roa, Trends Genet., 2010, 26, 443–448 CrossRef CAS PubMed.
- J. M. Soler, E. Artacho, J. D. Gale, A. García, J. Junquera, P. Ordejón and D. Sánchez-Portal, J. Phys.: Condens. Matter, 2002, 14, 2745–2779 CrossRef CAS.
- J. Ferrer, C. J. Lambert, V. M. García-Suárez, D. Z. Manrique, D. Visontai, L. Oroszlany, R. Rodríguez-Ferradás, I. Grace, S. W. D. Bailey, K. Gillemot, H. Sadeghi and L. A. Algharagholy, New J. Phys., 2014, 16, 093029 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8cp05991h |
| This journal is © the Owner Societies 2019 |