
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStability and behaviour in aqueous solutions of the anionic cubic silsesquioxane substituted with tetramethylammonium†
Emelie J.
Nilsson
 *a,
Marina
Huber
a,
Göran
Carlström
b,
Olle
Söderman
a,
Daniel T.
Bowron
c,
Karen J.
Edler
ad and
Viveka
Alfredsson
a
*a,
Marina
Huber
a,
Göran
Carlström
b,
Olle
Söderman
a,
Daniel T.
Bowron
c,
Karen J.
Edler
ad and
Viveka
Alfredsson
a
aPhysical Chemistry, KILU, Lund University, P.O. Box 124, SE-22100 Lund, Sweden. E-mail: nilsson.emelie.j@gmail.com
bCentre for Analysis and Synthesis, Lund University, P.O. Box 124, SE-22100 Lund, Sweden
cISIS, Science and Technology Facilities Council Rutherford Appleton Laboratory, Harwell Oxford, Didcot OX11 0QX, UK
dDepartment of Chemistry, University of Bath, Claverton Down, Bath, BA2 7AY, UK
First published on 4th March 2019
Abstract
The aqueous behaviour of the anionic octa-tetramethylammonium substituted cubic silsesquioxane, [N(CH3)4]8[Si8O20], was studied with quantitative 29Si-NMR. This molecule partially fragments in aqueous solutions, forming several smaller entities. The most abundant silica species are the monomer, dimer, cyclic trimer, cyclic tetramer and double three-ring. Higher concentrations are required in order to prevent complete fragmentation of the cubic structure. Additives such as alcohols and tetraalkylammonium salts have a stabilising effect on the cubic silsesquioxane, unlike sodium salts that destabilise it. A high concentration solution, containing the non-fragmented molecule as well as entities resulting from fragmentations, was investigated with neutron scattering coupled with modelling, using empirical potential structure refinement (EPSR). The modelling reveals that TMA+ ions coordinates to all different silica species, with approximately three TMA+ per cube. These are located at the faces of the cube.
Introduction
Silsesquioxanes are a group of silicon oxide based compounds, SiO3/2, typically including an organic moiety, R-SiO3/2. Silsesquioxanes often form discrete cage-like oligomeric structures, [R-SiO3/2]n, with n often being 6 or 8. For n = 8 a cubic structure is found, R8-Si8O12. This species has been thoroughly studied and can be functionalised by changing the organic moieties located at the corners.1 This cubic molecule is used in a wide variety of materials, ranging from nanocomposites for biomedical applications,2 flame-retardant polymers3 to films with low-dielectric constants.4 The cubic structure is interesting, not only from its perspective as an inorganic–organic hybrid material, but also due to its similarity to the double four membered ring found in many zeolite structures, such as zeolite A (LTA).5The cubic silsesquioxane can be substituted as a completely inorganic silicate salt, Si8O208−, with tetramethylammonium as counterions.6–9 This anionic cubic octamer is also observed, to a larger extent than smaller multiatomic silicate anions, during the formation of silica nanoparticles using tetraethoxysilane as the silica source.10,11 Silicate anions, present in aqueous solutions during silica polymerisation processes, have been extensively studied using 29Si-NMR spectroscopy10–20 and their expected 29Si-NMR chemical shifts have been the subject of calculations.21 Other smaller silicate species that have been identified to be present during the polymerisation process of silica materials10,11,15 include the monomer (silicic acid, Si(OH)4), the dimer (Si2O(OH)6), the cyclic trimer (Si3O3(OH)6, the cyclic tetramer Si4O4(OH)8) and the prismatic hexamer (double 3-ring, Si6O9(OH)6). The latter are often referred to as single 3-rings (S3R), single 4-rings (S4R) and double 3-rings (D3R) respectively. Using the same terminology, the cubic octamer is often referred to as the double 4-ring (D4R) and we will henceforth use this terminology. The different silica species are depicted in Fig. 1, along with symbols used to represent them. The D3R is also similar to the polyhedral oligomeric silsesquioxane, where n = 6. As both the D4R and the D3R are present in many polymerisation processes these species have been subjected to modelling.22–24 The models indicate that the D3R is less stable in solution than the cubic octamer. The cubic octamer has been suggested to be stabilised, and protected from hydrolysis, by association of tetramethylammonium ions (TMA+) to the faces of the D4R. This arrangement of the TMA+ ions allows the silanol groups of the D4R to interact with water while the more hydrophobic siloxane bridges remain protected from hydrolysis and subsequent polymerisation.
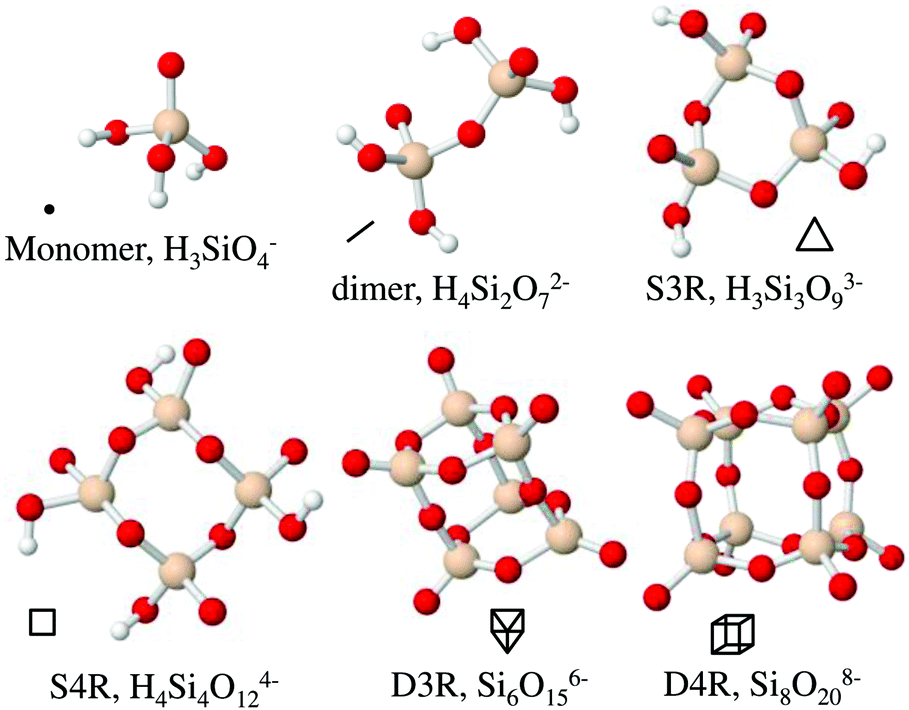 | ||
| Fig. 1 The six most frequently occurring silica species in solutions along with their acronyms and symbol representation. Beige spheres correspond to silicon atoms, red to oxygen atoms and white to hydrogen atoms. | ||
As the D4R is an essential secondary building unit in many zeolite structures there have been efforts to use the D4R in the synthesis of ordered mesoporous silica, replacing the frequently utilized silica source, tetraalkoxysilane (Si(OR)4).25 Mesoporous silica, a material ordered in the meso-regime (2–50 nm),26 is typically amorphous on the atomic level27 but using the D4R as a silica source suggests that local ordering could potentially be introduced. However, when the D4R (H8Si8O12) is used as silica source it fails to retain its structure and forms only a partly crystalline, or a completely amorphous, silica network.
In this study we aim to better understand the behaviour of small silica oligomers in aqueous solution primarily in order to shed light on the synthesis of porous silica materials. In the synthesis of zeolites, the synthesis relies on the interaction between polymerising siliceous species and molecular structure directing agents (SDAs), while in the synthesis of mesoporous materials the interactions between polymerising siliceous species and polymer/surfactant micelles are the driving force for formation. The typical silica source, tetraethylorthosilicate (TEOS), undergoes hydrolysis followed by polymerisation in solution, resulting in a highly dynamic system unsuitable for investigating molecular interactions. The D4R on the other hand could function as a model for a silica oligomer provided it is stable on the time scale of the measurements.
We have attempted to create an equilibrated system (or at least a system that is non-reactive during the duration of the measurements) that is not disturbed by the actual particle formation process, i.e. the silica polymerisation. The system contains the cubic silsesquioxane (D4R) with tetramethylammonium (TMA+) as counterion; Si8O20[N(CH3)4]8 (the crystalline form contains 65 water molecules9). We have investigated this salt in aqueous solutions, as a function of concentration and with addition of simple alkali salts, tetraalkylammonium salts or short chain alcohols. The particular concentrations of the D4R and the different additions are related to synthesis conditions of porous materials, either zeolite synthesis or the synthesis of mesoporous silica, or both. The salts that were added are sodium salts, with Cl−, Br− or I− as counterion, or tetraalkylammonium bromide, with one to three carbons in the alkyl group. For the tetramethylammonium ion, two additional counterions, Cl− or OH−, were also investigated. The effect of the sodium ion on the silica species is relevant as sodium silicate is another frequently used silica source. In addition, sodium salts have been shown to affect mesoporous silica materials, both for the non-ionic SDA system,28 as well as for the cationic SDA system.29 From a zeolite synthesis perspective, the interactions with tetraalkylammonium ions are of importance as these ions play a role as SDAs, and the smallest one, tetramethylammonium, has been shown to stabilise the D4R.6,20 Short chain alcohols, methanol and ethanol, are always present in a synthesis based on tetraalkoxysilanes; during hydrolysis, alcohol is produced in a molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 for Si:alcohol.
:4 for Si:alcohol.
The main tool of our investigation is quantitative 29Si Nuclear Magnetic Resonance (NMR) spectroscopy. NMR spectroscopy allows identification and quantification of the siliceous components as well as insight to the stability of the cubic octamer in the different aqueous environments. The NMR investigations are complemented with neutron scattering and empirical potential structure refinement (EPSR) modelling30–33 for one concentration of the silica salt in water, which provides an atomistic description of the cubic octamer and its environment.
Materials and methods
The anionic octa-tetramethylammonium substituted cubic silsesquioxane, [N(CH3)4]8[Si8O20]·xH2O, (PSS hydrate-octakis(tetramethylammonium) substituted), bought from Sigma Aldrich, was used as received. Tetramethylammonium bromide (Sigma Aldrich, >98%), chloride (Acros Organics, >98%) and hydroxide (Sigma Aldrich, 25 wt% in H2O), tetraethylammonium bromide (Sigma Aldrich, >98%), tetrapropylammonium bromide (Sigma Aldrich, >98%), NaCl (Merck, min 99.5%), NaBr (Prolabo, min 99%) and NaI (AppliChem, min 99.5%) were all used as received. D2O was bought from Sigma-Aldrich (99.9 atom% D), and all H2O was ultrapure Merck Millipore water (resistance of 18.5 MΩ cm). After weighing the additives into glass vials, and dissolving it in the solvent, the silica salt i.e. the cubic silsesquioxane, was added to the solutions by weight. The vials were then gently shaken until the salt dissolved. All samples were measured immediately, unless otherwise specified.The pH-measurements were performed using a PHM210 Standard pH Meter, MeterLab from Radiometer analytical connected to a Blue Line 17 pH electrode from SI analytic. The measurements were performed at room temperature with the pH meter calibrated to two standard buffer solutions (pH 7.00 (20 °C) and pH 10.00 (20 °C)) from ScharLab. All samples were measured immediately after preparation, and again after one month at room temperature. No changes to the pH values were observed in this time span, and so effects due to dissolved CO2 in the samples were not taken into account.
The water content in the silica salt, [N(CH3)4]8[Si8O20]·xH2O, was quantified by thermogravimetric analysis (TGA) using a Q500 Analyzer (TA Instruments). The salt was subjected to a heating rate of 10 °C min−1 with a nitrogen flow of 60 ml min−1 over the sample. The samples were heated from room temperature to 600 °C in aluminium pans. The data, found in the ESI,† Fig. S1, is in good agreement with published TGA studies of this hydrated salt.34 The salt contained 53.2 wt% water, i.e. [N(CH3)4]8[Si8O20]·72H2O, which is in good agreement with the previously found 65 crystal waters by X-ray diffraction.9 TGA data for the salt from a newly opened bottle and a bottle opened two years earlier (then left standing, closed and wrapped with parafilm, on the shelf, but exposed to normal humidity and temperature fluctuations) showed identical results (Fig. S1, ESI†). Hence, the salt showed no tendency to absorb more water over time.
The 29Si(1H)-NMR experiments were performed with proton decoupling only during the acquisition time, providing decoupled spectra without any nuclear Overhauser effect (NOE). The measurements were carried out at room temperature, 25 °C, on a Varian UNITY Inova 500 MHz spectrometer at a resonance frequency of 99.28 MHz using a 5 mm Varian Nalorac Z·SPEC Dual Broadband Gradient probe (DBG500-5EF). 128 scans were acquired in all spectra using a pulse length of 8.3 μs (90°), an acquisition time of 3 s, and a relaxation delay of 82 s, giving a total inter pulse delay of 85 s, corresponding to five times the longitudinal relaxation time (T1) for the slowest relaxing species. Each spectrum required about three hours of measurement time. 29851 complex data points were collected, using a spectral width of 9950 Hz (100.2 ppm). All samples were measured in water solutions containing 10 wt% D2O for the spectrometer lock, with an inner capillary containing pure methyltrimethoxysilane as an external reference for the chemical shifts. All acquired data were Fourier transformed with ACD/NMR Processor (Academic Edition) using an exponential window function with a line broadening of 1 Hz and zero filling to 128k data points. The relative concentrations of the different silica species were determined by numerical integration of the corresponding resonance peaks using the ACD/NMR Processor, after phase and base line correction. All peaks are referenced relative to the external reference methyltrimethoxysilane at −40.44 ppm. The chemical shift of −40.44 ppm was experimentally referenced to tetramethylsilane (TMS) at zero ppm, by putting the inner capillary of methyltrimethoxysilane in a sample of pure TMS. To assess the variation in the results, different preparations of 6 and 22 wt% silica salt solutions were measured.
Longitudinal relaxation times, T1, seen in Table 1, were measured using the inversion recovery method with relaxation delays in the range of 0.003–60 s, and a recycle delay of 110 s between each scan to allow for full recovery of the magnetization. The single exponential fits to the data are shown in ESI,† Fig. S2.
| Relaxation time, T1 | Monomer | Dimer | S3R | S4R | D3R | D4R |
|---|---|---|---|---|---|---|
| T 1 [s] | 16.4 | 15.8 | 16.4 | 16.0 | 9.1 | 5.2 |
The solutions for the neutron scattering experiments were mixed at three different isotopic compositions; H2O, D2O or a 50:50 molar ratio of H2O:D2O (HDO). A solution containing 22 wt% of the silica salt was investigated. The three contrasts were measured at the near and intermediate range order diffractometer NIMROD at STFC's ISIS Neutron Scattering Facility, UK.35 A Q-range of 0.02–50.0 Å−1, corresponding to a distance between 0.1 to 300 Å, is achievable at NIMROD. Q is the magnitude of the scattering vector, Q = (4π/λ)sin(θ), where 2θ is the scattering angle and λ is the wavelength of the beam. 1.5 ml of each sample was prepared for the 1.4 ml flat plate cells used for the experiment. The cells have a 1 mm slit opening and 1 mm thick walls constructed of null-scattering TiZr-alloy walls. The cells were exposed to a rectangular beam (30 × 25 mm, H × V) under vacuum, until 80 μA beam current was reached (roughly corresponding to two hours). An automatic sample changer allowed multiple cells to be handled simultaneously in the sample chamber. Due to the possibility of loading multiple samples simultaneously, the time between mixing and measuring was occasionally as long as three days. The cells were weighed before and after measurements to verify that no leakage had occurred during the measurement. The samples were measured at 25 °C. Additionally, the empty instrument and a vanadium plate (applied as a standard) were measured equivalently to the samples. The Gudrun program36 was used for data correction and normalisation.
Empirical potential structure refinement,30–33 EPSR, was used to model the neutron scattering data. EPSR applies an empirical potential to a reference system, a classical pair potential based on Lennard-Jones potential and Coulomb interactions. The parameters used for the modelled silica species were based on literature data.22,23,37 The counterion, tetramethylammonium (TMA+), was modelled according to previously published work,38 and water was based on the SPC/E model. All parameters are presented in the ESI,† Table S1. The model is consistent with 22 wt% silica salt in water, measured using neutron scattering, and is constrained by chemical and physical parameters (e.g. the density (0.1 atom Å−3), molecular structures and atomic overlaps). The number of species in the model for the 22 wt% sample was 28 monomers, 6 dimers and 14 D4Rs to 11248 water molecules which corresponds to a Si:TMA+:H2O ratio of 1:1:74. The ratio of the species was determined by 29Si-NMR, see below. The cubic simulation box had a length of 71.8 Å. The model based on the reference potential, starting from a random configuration, was allowed to reach equilibrium at which point the empirical potential was applied. The system was again allowed to reach equilibrium, and at least 25000 EPSR cycles were accumulated.
Result and discussion
Identification of species using 29Si-NMR spectroscopy
The 29Si-NMR spectroscopy reveals that the D4R in solution fragments into a range of different silica species, see Fig. 2. This process occurs quickly, prior to the first measurement. The first acquired NMR spectrum indicates results analogous to those acquired at the end of the measurement. Each individual scan of the 29Si-NMR spectra exhibits analogous intensities, for each sample, during the measurement (3 hours, i.e. 128 scans). In addition, a freshly prepared sample revealed the same NMR results as that of a sample dissolved one week before (29Si-NMR spectra can be found in Fig. S3 in ESI†). No apparent silica polymerisation, or changes in concentration of the different silica species, was detected, during the time frame of our investigation (except the initial fragmentation occurring on mixing, i.e. prior to all measurements). Note that the neutron measurements were performed on samples not older than one week. Hence, for the purpose of this study we consider the system to be in a temporary steady state. However, if left over a period of several months, the silica species will polymerise.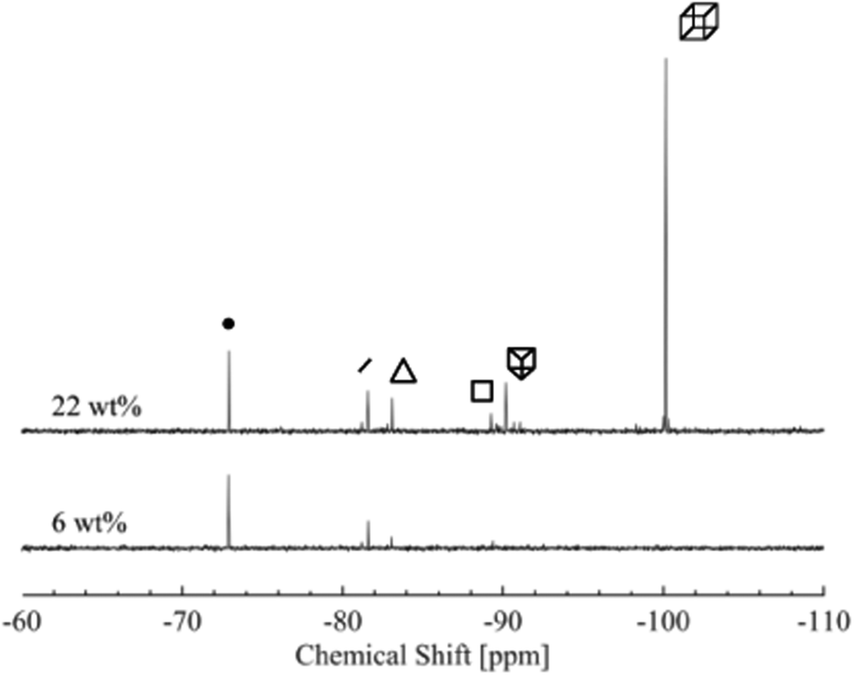 | ||
| Fig. 2 29Si-NMR spectra of two concentrations of the silica salt, 6 and 22 wt%. Each of the main species present in the solution is indicated by its corresponding symbol, see Table 2. | ||
Aqueous silicate species have over the last decades been extensively studied and characterised with 29Si-NMR spectroscopy.12,16,17,40 The 29Si-NMR spectra typically reveal six peaks, which were assigned to the monomer, dimer, S3R, S4R, D3R and D4R, respectively. See Table 2 for chemical shifts of the most abundant species, obtained in this study, together with values from the literature.16,17,19 The peaks at more negative chemical shifts gradually decrease in intensity and eventually disappear as the silica salt concentration decreases (Fig. S4 and Table S2 in the ESI†).
| Species | Exp. δa [ppm] | δ [ppm]19 | δ [ppm]16 | δ [ppm]39 | Symbol |
|---|---|---|---|---|---|
| a From experiments, at 12 wt% of silica salt, corresponding to the lowest concentration with all species present. | |||||
| Monomer | −72.88 | −72.26 | −71.00 | −73.10 | ● |
| Dimer | −81.59 | −80.94 | −79.62 | −79.92 | — |
| S3R | −83.07 | −82.37 | −81.11 | −81.49 |

|
| S4R | −89.29 | −88.60 | −87.04 | — |

|
| D3R | −90.13 | −89.32 | −88.06 | −90.40 |

|
| D4R | −99.89 | −98.99 | −98.12 | −99.30 |
|
An important parameter for a silica material synthesis is the pH value as it will, for instance, have a large impact on the polymerisation rate. The pH was measured, but not actively adjusted, in these samples. It was noted that after addition of the silica salt to pure water the pH increased, see Table 3 and Table S3 (ESI†). All samples containing additional sodium or tetraalkylammonium salts (TMAOH excluded), retained a pH analogous to that of a sample containing only silica salt. The increase in pH therefore appears to be related to the fragmentation of the D4R, and consequently the amount of added silica salt. If the D4R stays intact, but completely protonates, an increase in pH will occur according to the reaction below:
| Si8O208− + 8H2O ⇌ Si8O12(OH)8 + 8OH− |
| Si8O208− + 20H2O ⇌ 8Si(OH)4 + 8OH− |
| Weight% silica salt | Si atom concentration [M] | pH | Exp. δ [ppm] of D4R |
|---|---|---|---|
| 3 | 0.096 | 11.53 | — |
| 6 | 0.195 | 11.85 | — |
| 12 | 0.390 | 12.11 | −99.89 |
| 18 | 0.585 | 12.26 | −100.06 |
| 22 | 0.736 | 12.32 | −100.14 |
| 29 | 0.928 | 12.34 | −100.20 |
Quantification of the relative concentrations of the main silicate species from the 29Si-NMR spectra (Fig. S4, ESI†), shows that there is a concentration dependence of the different species, see Fig. 3. The D4R is the dominant species only for the higher concentrations and appears absent below 6 wt%. For the lower concentrations, the monomer is by far the most abundant species. The other four species (dimer, S3R, S4R and D3R) are less dependent on the total silica concentration in the solution; their respective concentrations are almost constant. The D3R appears to be less stable, or less prone to form, in the aqueous solutions compared to the D4R, in agreement with previous studies of the formation of silica oligomers.10,11,14,42 The concentrations of these four, almost constant, species are always low, in contrast to the D4R or the monomer. The variations in the results can be seen in Fig. 3 for the 6 wt% and 22 wt% sample. The variation is larger for the lower concentrations. A low concentration leads to a low signal-to-noise ratio in the 29Si-NMR spectrum, making it more difficult to accurately determine the concentrations of these species. Nevertheless, the measurements of different samples reveal only a small spread between the measured concentrations. A number of other peaks were also detected in the samples, see Fig. 3 (Fig. S4 and S7, ESI†). These peaks have previously been subjected to investigation and are all species similar to the main species, but are not symmetrical species. This will give rise to multiple peaks.43,44 Due to their low abundance (at most only 9% of the Si-atoms in the samples, spread over 5–14 different peaks) these peaks were not assigned as it was considered outside the scope of this paper.
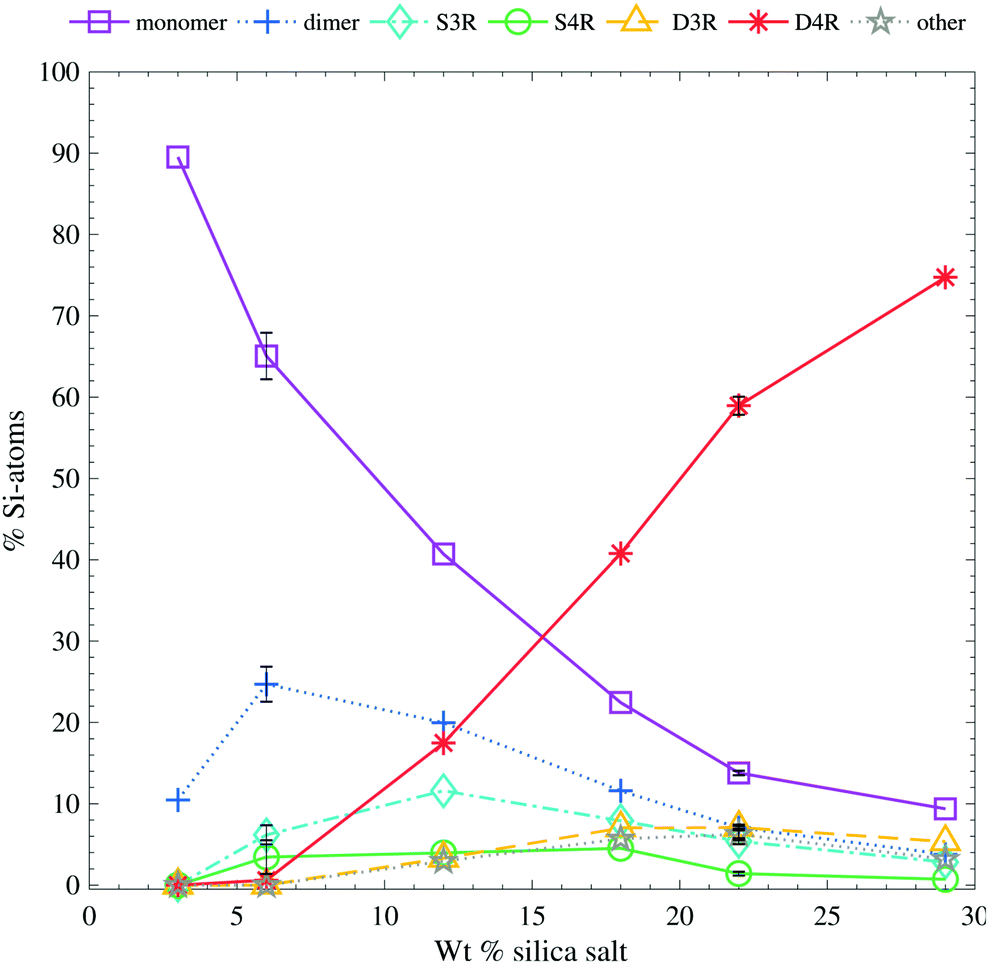 | ||
| Fig. 3 The relative Si-atom concentration of the main silica species, obtained from the 29Si-NMR, as a function of silica salt addition (as follows: monomer purple squares, dimer blue crosses, S3R teal diamonds, S4R green rings, D3Rs yellow triangles, D4Rs red stars and other assorted species grey pentagrams), the line is drawn as a guide for the eye (see Table S2 in ESI† for all values). The relative Si-atom concentration is the percentage of the total number of Si atoms in the sample. (e.g. a ratio of the total number of Si-atoms in the dimer divided by the total number of Si-atoms in the sample). The error bars are one standard deviation obtained from 6 and 4, repeated measurements of the 6 wt% and 22 wt% samples, respectively. The corresponding 29Si-NMR spectra are shown in Fig. S4 (ESI†). | ||
The D4R appears to be unstable below a certain silica concentration. A similar behaviour has been observed by Hasegawa et al.6 during syntheses of the D4R, in the presence of TMA+. They did not observe any D4R below a starting concentration of 0.3 M SiO2 (cf.Table 3 for the concentrations used in this study). This is in line with the suggestion that TMA+ stabilises the D4R but, at lower concentrations, due to entropic reasons, TMA+ does not associate with the D4R and hence no D4R are present.
It has been reported that below 0.7:1 the OH−:Si ratio affects the stability of aqueous silicates in tetraalkylammonium solutions, and due to polymerisation colloidal silica starts to form.14 This is observed as a broadening of the Q2, Q3 and Q4 resonances in 29Si-NMR (Qn is used to denote the resonance corresponding to the Si-atom linked to n other Si-atoms, e.g. the monomer will be Q0). Broadening was not observed in any of our NMR spectra, measured within a week of preparation, despite the fact that the OH−:Si ratio is below 0.7 for the three lowest concentrations (0.2, 0.3, 0.4 respectively with increasing Si, from calculations based on the pH measurements without taking dissolved CO2 into account). The three highest concentrations have a ratio larger than 0.7 (1.0, 1.1, 1.1 respectively). Despite our systems’ OH−:Si ratios, overall silica polymerisation is considered to be absent within the time frame of our experiments. Hence the systems are considered to be in a temporary steady state.
A small upfield change of the 29Si-NMR chemical shifts with increasing concentration is observed for the D4R, see Table 3 (observable in all repetitive measurements). Similar changes in the chemical shift have previously been reported in literature for both tetraalkylammonium and alkali silicate solutions.15,18,42 This effect was attributed to a change in the degree of protonation and/or to the degree of association of cations, as well as to variations in temperature. Similar changes in the chemical shifts was also reported for other silicate species,15,18,42 but to a smaller extent. In one of these studies, the shifts appeared more dependent on the temperature and identity of the cation rather than degree of protonation.42 Since the cation in the investigated system, its relative concentration (1 TMA+:1 Si), and the temperature are all kept constant here, the main change in chemical shift could be attributed to the degree of protonation. The observed shift corresponds well with the adjustment of number of charges related to D4R in the solution (as found by the pH measurements). However, this upfield change is most likely dependent on several parameters, and cannot be used to derive the degree of protonation of the D4R with any certainty.
Addition of TAA+ ions
Addition of tetraalkylammonium ions, regardless of the alkyl chain length or anionic counterion, increases the stability of the cubic octamer, Fig. 4. Addition of 0.1 M TAA+ to a 6 wt% salt sample (a TAA+:Si ratio of 1.5:1.0 including the inherent TMA+ counterion), is sufficient to start stabilising the D4R. The degree of stabilisation reveals no, or only a small, correlation to the anionic counterion of the TAA+ salt or to the alkyl chain length, see Fig. 4. This was unexpected as it has previously been reported that the D4R was destabilised when TEA+ was added to a TMA+ stabilised D4R solution.14 Further, that study also showed a decrease in the D4R concentration, in the syntheses of silicate species, as the alkyl chain length of the TAA+ was increased.20 This was attributed to the decreased charge-to-radius ratio of the longer alkyl chain length TAAs. Tetramethylammonium ions are small enough to effectively screen the charges and allow the silanols to interact with water, while at the same time are big enough to protect the D4R against hydrolysis. The TPA+ ions are more hydrophobic than TMA+ ions in water,38 which could influence their stabilising ability. At higher concentrations, TPA+ ions may favour interactions with each other rather than with the more polar silica species, leading to a less efficient stabilising ability compared to TMA+. However, the data here shows that the increased TAA+:Si ratio is sufficient to enhance the stability of the D4R. At these overall low TAA+ concentrations pairing or clustering of these ions are not expected. A small decrease in the monomer concentration, and increase in mid-sized species, is noticed for the TMAOH addition.
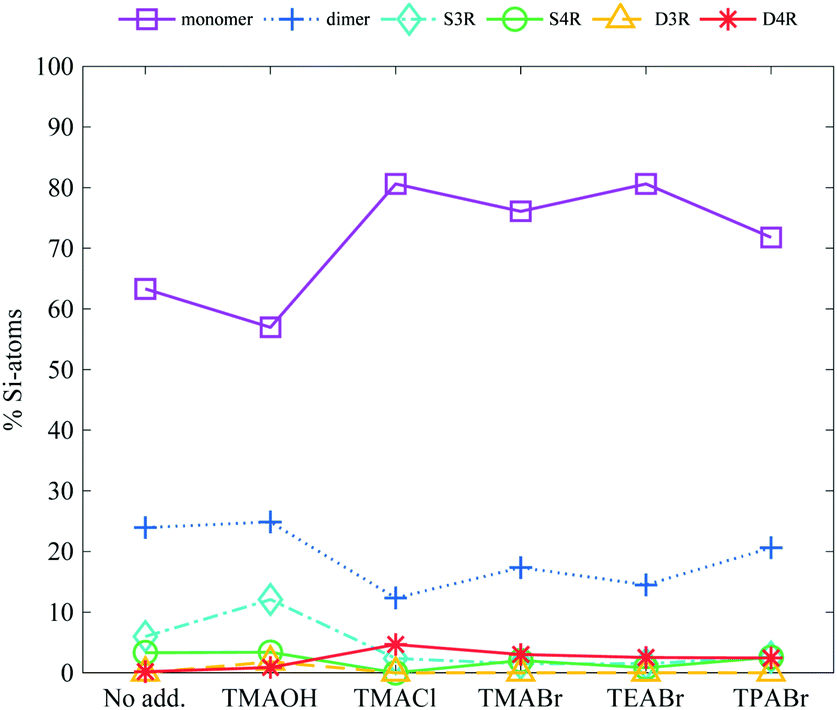 | ||
| Fig. 4 The relative Si-atom concentration of the main silica species, obtained from the 29Si-NMR, of a 6 wt% silica salt sample with addition of 0.1 M TAA-salts (as follows: monomer purple squares, dimer blue crosses, S3R teal diamonds, S4R green rings, D3Rs yellow triangles, D4Rs red stars and other assorted species grey pentagrams), the line is drawn as a guide for the eye (see Table S2 in ESI† for all values). | ||
Addition of sodium salts
Additions of alkali salts have previously been reported to destabilise the D4R14,15 and when TAA+ ions are exchanged for alkali ions in a synthesis, mid-sized silicate species are favoured.20 This destabilising behaviour was credited to smaller cations being able to expel TMA+ ions from the sides of the D4R, leaving the D4R vulnerable to hydrolysis.As expected the addition of sodium salts, 0.1 or 0.2 M (a Si:TMA+:Na+ ratio of 1:1:0.5 and or 1:1:1 respectively, with TMA+ being the inherent counterion), does not stabilise the D4R at a concentration of 6 wt% silica salt, see Fig. 5. Instead it caused a small increase in the monomer concentration, and a decrease in the dimer concentration. If TMA+ associated to all sides of the D4R, at an addition of 0.2 M Na-salts all TMA+ ions could be replaced by Na+, given it is a favourable exchange. This study confirms that the Na+ ion generates no observable stabilisation of the D4R, but rather seems to enhance the fragmentation. This is observed as a small increase in the monomer concentration and a decrease in the dimer concentration. Exchanging the anionic counterion (Br−, Cl− or I−) showed no signs of influencing the silica speciation.
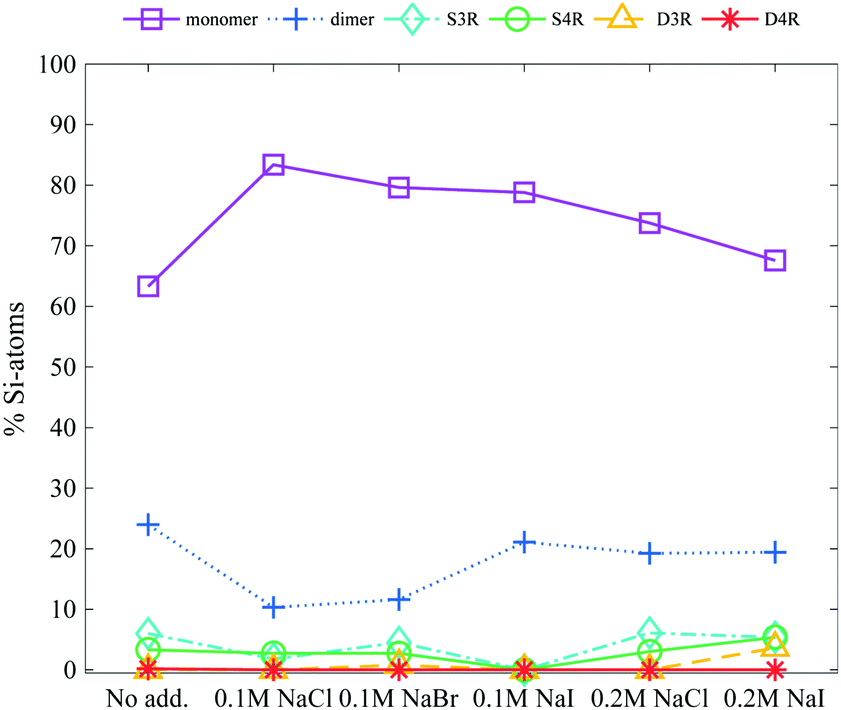 | ||
| Fig. 5 The relative Si-atom concentration of the main silica species, obtained from the 29Si-NMR, of a 6 wt% silica salt sample with addition of 0.1 M or 0.2 M Na-salts (as follows: monomer purple squares, dimer blue crosses, S3R teal diamonds, S4R green rings, D3Rs yellow triangles, D4Rs red stars and other assorted species grey pentagrams), the line is drawn as a guide for the eye (see Table S2 in ESI† for all values). | ||
Addition of short chain alcohols
In a silica synthesis, using alkoxysilane as the silica source, four alcohol molecules are produced per silicon atom in the hydrolysis. To focus on the effect of addition of short chain alcohols to a silica synthesis (using an alkoxysilane as the silica source) an alcohol concentration was chosen so that the Si:alcohol ratio was 1:8.5. Ethanol and methanol additions had a large impact on the stabilisation of the D4R, see Fig. 6. Alcohol addition stabilised the D4R even at low silica salt concentrations (6 wt%). At the higher concentration, 22 wt% silica salt, the majority of the Si atoms remained in the form of D4Rs, consequently resulting in lower concentrations of monomers and dimers. Both ethanol and methanol greatly stabilised the D4R, however, to different extents; ethanol is found to be a more efficient stabiliser.
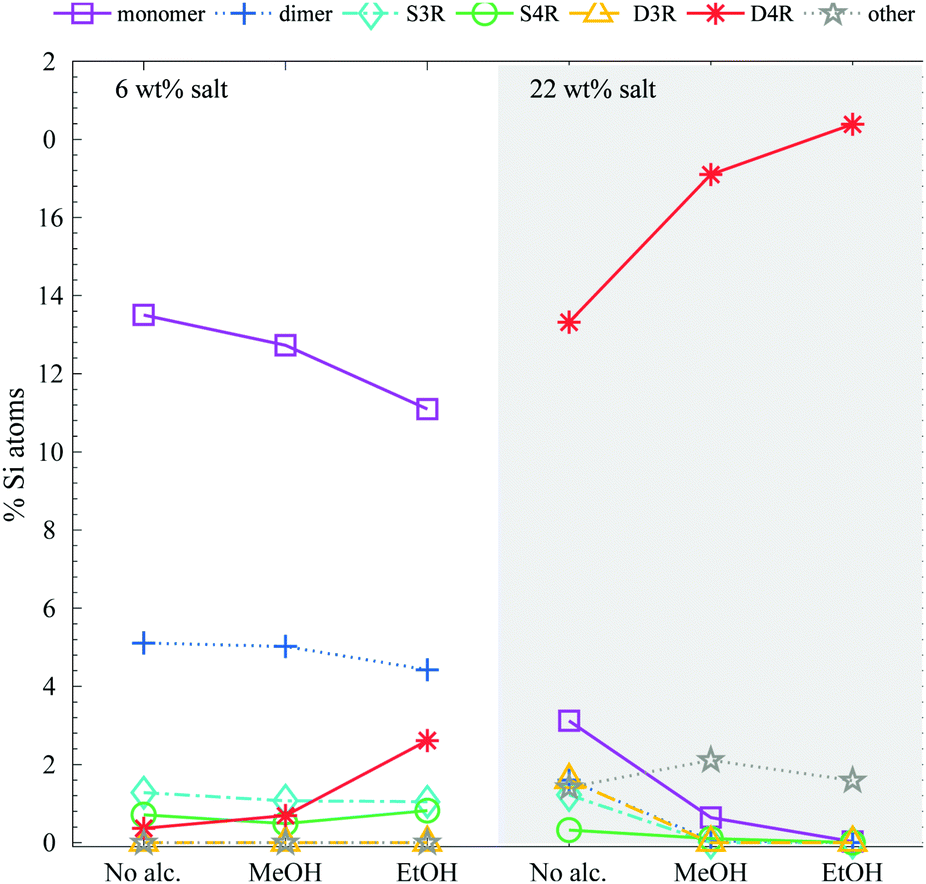 | ||
| Fig. 6 The relative Si-atom concentration of the main silica species, obtained from the 29Si-NMR, of a 6 and 22 wt% silica salt sample with added methanol or ethanol to a ratio of 1:8.5 Si:alcohol (as follows: monomer purple squares, dimer blue crosses, S3R teal diamonds, S4R green rings, D3Rs yellow triangles, D4Rs red stars and other assorted species grey pentagrams), the line is drawn as a guide for the eye (see Table S2 in ESI† for all values). The corresponding 29Si-NMR spectra are shown in Fig. S7 (ESI†). | ||
Hasegawa et al.6 found that methanolic solutions were beneficial as compared to aqueous solutions when synthesising D4R, and if above a specific methanol concentration the monomer was no longer observed. This was explained to be a consequence of cosolvents decreasing the water activity, promoting ion association and as a consequence even the larger TAA+ ions stabilised the D4R during synthesis.14
Another aspect of the alcohols is their “amphiphilic” behaviour, having polar OH groups and apolar alkyl chains, a behaviour they share with the TAA+ ions. It appears as if species with an amphiphilic character protect the D4R from hydrolysis. Possibly, the apolar part interacts with the more hydrophobic siloxane bridges of the D4R, whereas the polar part interacts with water or the charged moieties.
Another possibility is that the alcohol binds through an ether bond to the corners of the D4R, as in the above mentioned organic–inorganic hybrid materials. As the 29Si-NMR spectra were normally measured proton decoupled, this would not be observed in our study. However, if the proton decoupling is removed, a splitting of the 29Si resonance is expected due to the scalar coupling between the protons of the alcohol and the silicon nucleus.45 To rule out the formation of any ether bonds between the alcohol and the D4R, a coupled spectrum was obtained, see Fig. 7. No peak splitting was observed, i.e. the alcohol is not expected to bind to the D4R.
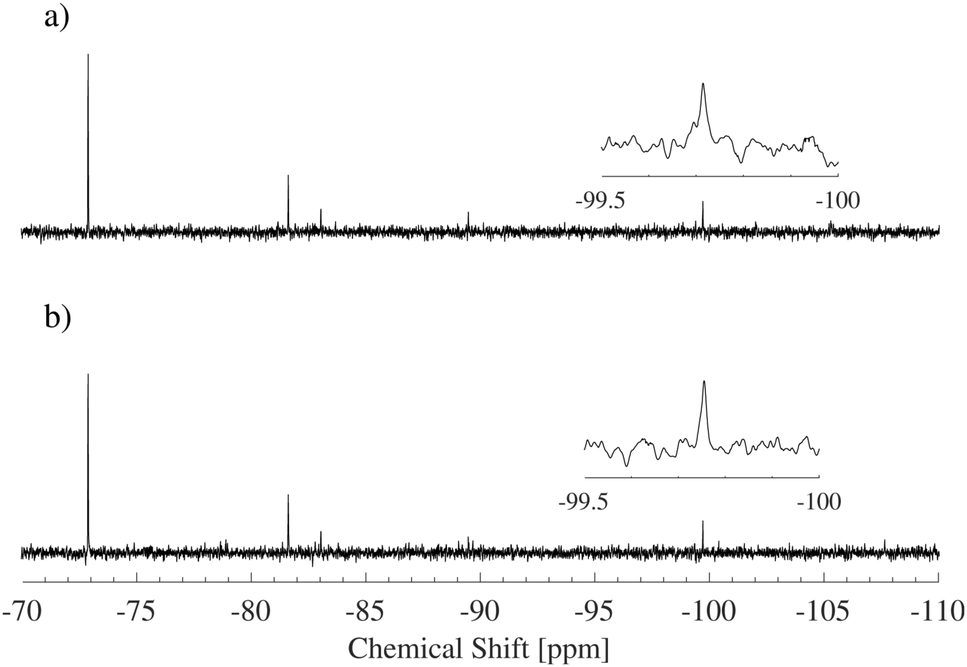 | ||
| Fig. 7 29Si-NMR spectra for a 6 wt% salt solution in 1.7 M EtOH, (a) 1H-decoupled and (b) without proton decoupling. If the alcohol had formed an ether bond with the cube, a splitting of the peak at −99 to −100 ppm in spectrum (b) would have been expected. | ||
EPSR modelling
Neutron scattering data was collected on solutions of 22 wt% D4R in aqueous solutions and analysed using EPSR modelling. To construct the EPSR models for each molecule, an individual charge was assigned to each atom. This defines the electrostatic reference potential for that atom. All silica species can act as mono- or multivalent acids. The number of negative charges required for electroneutrality was calculated for the 22 wt% solution, based on pH and considering the concentration of the respective ions in the solution. Assuming that each Si–OH has a pKa1 close to that of silicic acid, irrespective of the size of the molecule, it was assumed that all silica species had gone through one full protonation step. It was further assumed that the pH was sufficiently below pKa2 so that no further protonation occurred (see Table 3 and Table S3, ESI†). The molecules will thus have as many negative charges as it has Si-atoms, hence the monomer has one charge, the dimer two and the D4R eight. A pH change is known to happen when substituting hydrogen for deuterium, but a slightly lower pH is unlikely to affect the first protonation step. Hence, the assumption that all silica species have undergone a complete first protonation step is still valid. Due to the limitations of the box size, computational time and the corresponding statistics, the 22 wt% sample was approximated to contain only monomers, dimers and D4Rs. In mole% these species comprise of 72% of the species present (i.e. 88% of the total number of Si atoms), according to the NMR measurements. The above approximation would outweigh any small changes in speciation originating from a change in pH when substituting hydrogen for deuterium.The scattering data and the corresponding fits are displayed in Fig. 8, and each data set is offset along the ordinate for clarity. The system appears to have little effect on the behaviour of water examining the radial distribution functions for oxygen (OW) and hydrogen (HW), see Fig. S8 in ESI.† This is expected considering that the main constituent in the system is water. A comparison of the behaviour of the TMA+ ions, with that reported in previous work,38 shows that the hydration is similar to that of the same ion in solutions of the halide salt, TMABr (Fig. S9, ESI†); it retains a tetrahedral arrangement (Fig. S10, ESI†) and has a hydration number around 25, see Table 4. However, some structure in the water arrangement is lost in the presence of the silica species, which could be a consequence of TMA+ associating to the silica species, preventing the ion from retaining its full water arrangement.
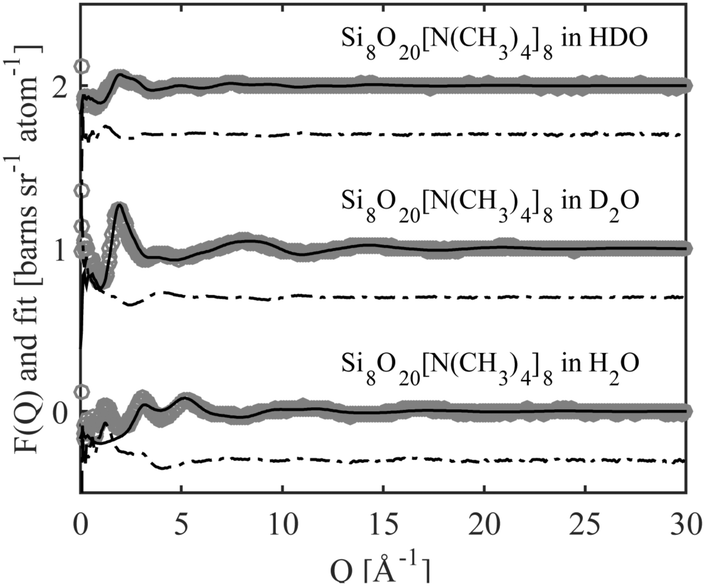 | ||
| Fig. 8 The neutron scattering data (grey circles), the EPSR fit (solid black line) and the difference between them (dot-dashed black line). The different systems have been offset along the ordinate for clarity. | ||
| Pair correlation | R min [Å] | R max [Å] | Nbr of atoms | Standard deviation |
|---|---|---|---|---|
| Simonomer–NTMA | 3.5 | 8.5 | 1.1 | ±0.8 |
| Simonomer–OW | 2.0 | 5.5 | 19.7 | ±2.1 |
| NTMA–OW | 3.0 | 6.0 | 25.5 | ±2.3 |
The site–site radial distribution functions, gab(r), show that the central nitrogen in the TMA+ (NTMA+) correlates strongly to the monomer's Si atom (Simonomer) and the interaction is mediated by protonated and deprotonated oxygens on the monomer, see Fig. 9. The first peak reveals that the monomer coordinates to approximately 1 TMA+, demonstrating that TMA+ is strongly associated with these silica species. The site–site radial distribution functions for the Simonomer to the oxygen (OW) and hydrogen (HW) of water reveals the strongly anionic nature of the monomer; the HW are turned towards the ion, see Fig. 10. The Simonomer coordinates to approximately 19 water molecules (Table 4). Fig. 11 displays the top 15% probability of locating OW and NTMA around Simonomer, based on calculated spatial density functions. The arrangement demonstrates that both water and TMA+ prefer the sites around the monomer where the Si atom is exposed (the tetrahedral faces). For water there is also a strong correlation around the deprotonated site of the monomer (one corner), whereas the probability of finding the TMA+ at the same location is much lower. This is not intuitive as TMA+ is expected to have an affinity for the highly negatively charge deprotonated site. However, the hydrophobic behaviour of the TMA+ possibly plays a role; TMA+ correlates more readily to the protonated oxygens, see Fig. 9 and 11.
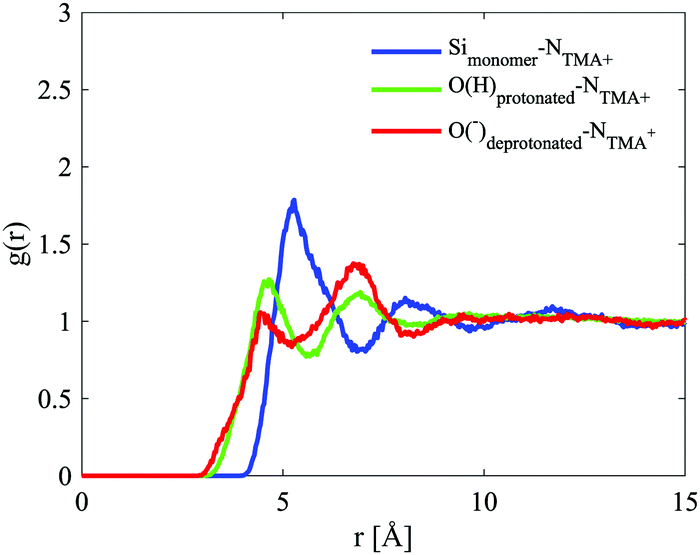 | ||
| Fig. 9 Site–site radial distribution functions for the monomer and the central nitrogen in TMA+ (Si–N in blue, Odeprotonated–N in red and Oprotonated–N in green). | ||
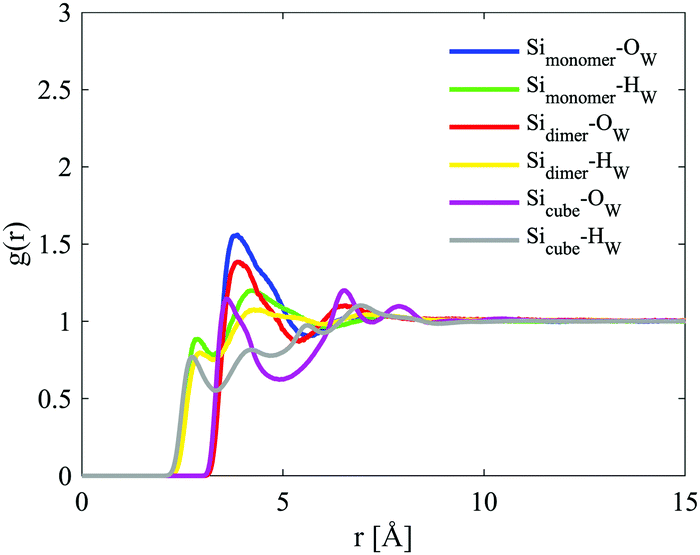 | ||
| Fig. 10 Site–site radial distribution functions for the Si atoms in the three different species, with the oxygen (OW) and hydrogen (HW) of water. The curves are depicted in different colours: Simonomer–Ow in blue, Simonomer–Hw in green, Sidimer–Ow in red, Sidimer–Hw in yellow, Sicube–Ow in magenta and Sicube–Hw in grey. | ||
 | ||
| Fig. 11 Spatial density functions showing, in blue, the top 15% for Simonomer to NTMA between 3 to 8 Å, and, in red, the top 15% for Simonomer and OW between 3 to 5.5 Å, from two directions. | ||
The site–site radial distribution for the NTMA+ to the oxygen atoms in the dimer shows, as for the monomer case, more distinct correlations to the protonated and deprotonated sites compared to the oxygen atom in the siloxane bridge. The dimer also exhibits, like the monomer, a water arrangement around the ion indicative of an anion (Fig. 12).
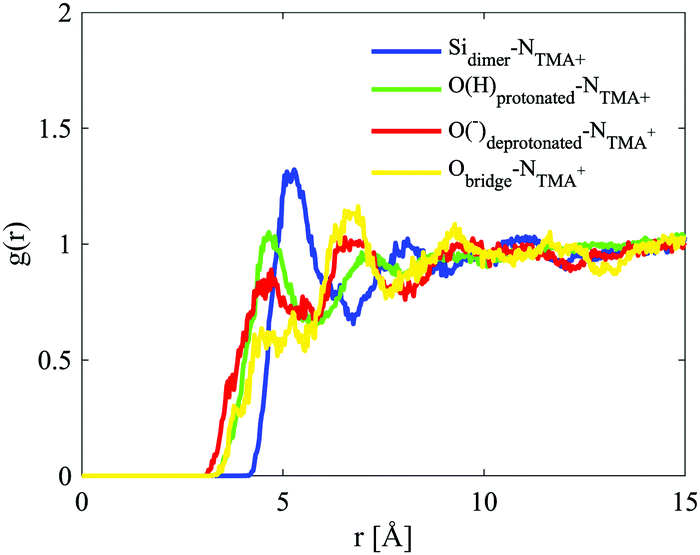 | ||
| Fig. 12 Site–site radial distribution functions for the dimer and the central nitrogen in TMA+. The curves are depicted in different colours: Sidimer–NTMA in blue, Oprotonated–NTMA in green, Odeprotonated–NTMA in red and Obridge–NTMA in yellow. | ||
This anionic character is also shared by the D4R; a character, therefore, present for all of the modelled silicate species. The D4R behaviour is equally attributed to water orientation in the vicinity of the cube, see Fig. 10 and 14.
For the D4R, the most pronounced correlation for the NTMA+ is, in contrast to both the dimer and monomer, to the deprotonated oxygen. The second strongest correlation is to the Si atom (Sicube), see Fig. 13, similar to the monomer and dimer. TMA+ has a weaker correlation to the oxygen in the siloxane bridge, which seems contradictory to TMA+ ion associating with the faces of the D4R. In a molecular dynamics study, of Si8O208− with TMA+ in solution, Caratzoulas et al.22 calculates the positions of TMA+ ions around the D4R. Their first peak in gSi–N(r) is located at 4.5 Å and the second at 7.7 Å. The assumption made is that the NTMA+ is positioned a distance out from the centre of the D4R faces, 4.5 Å from the closest Si atoms.
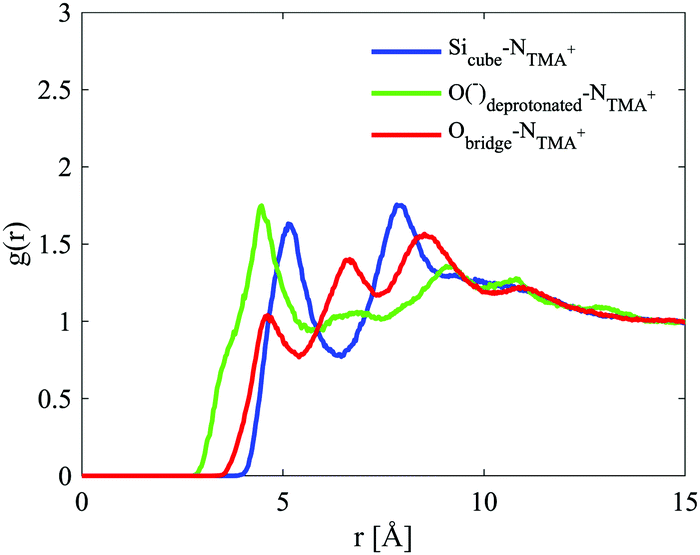 | ||
| Fig. 13 Site–site radial distribution functions for the cube and the central nitrogen in TMA+. The curves are depicted in different colours: Sicube–NTMA in blue, Odeprotonated–NTMA in green and Obridge–NTMA in red. | ||
TMA+ hence has an octahedral coordination around the D4R. Using the equation:
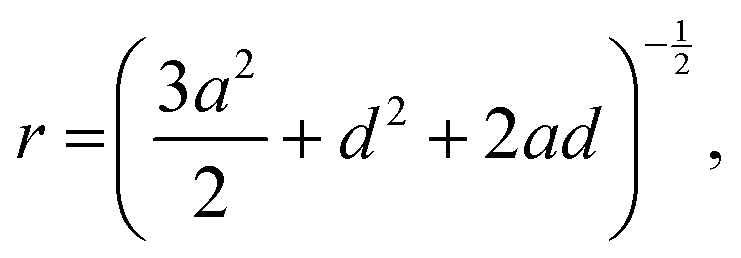
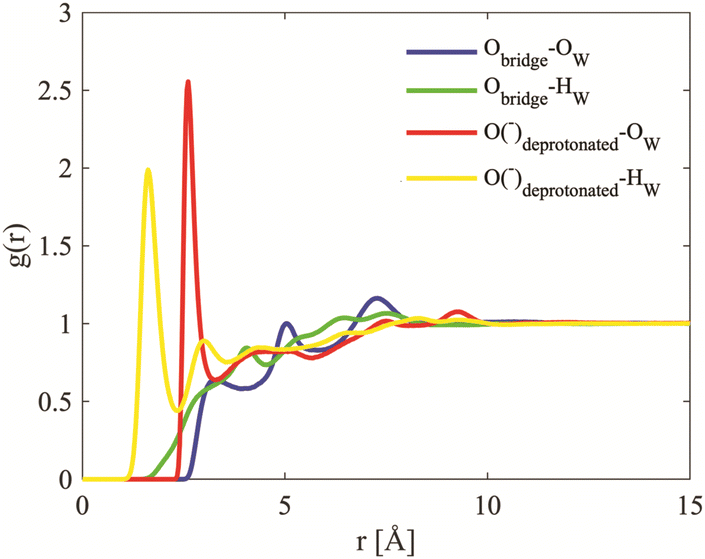 | ||
| Fig. 14 Site–site radial distribution functions for the oxygens in the cube and the oxygen hydrogen of water. The curves are depicted in different colours: Obridge–Ow in blue, Obridge–Hw in green, Odeprotonated–Ow in red and Odeprotonated–Hw in yellow. | ||
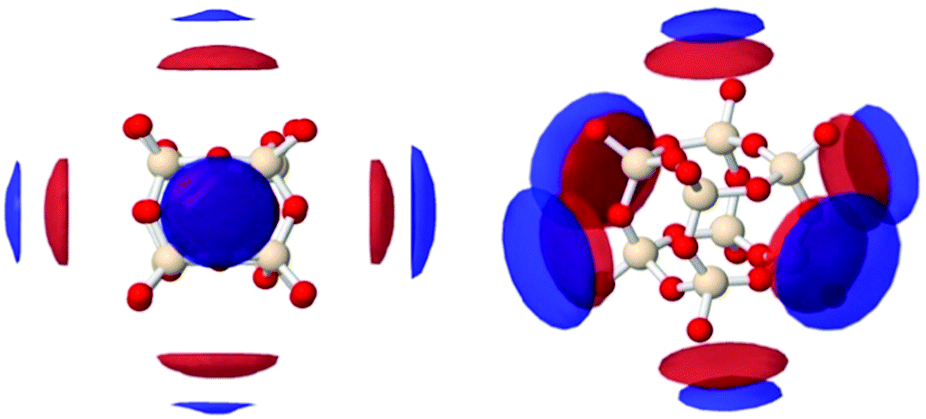 | ||
| Fig. 15 Spatial density functions (SDF) showing most probable locations of the oxygen of water (red) and the TMA+ nitrogen (blue, NTMA+) around the D4R. The SDFs show the top 15% of OW to Sicube between 2.0 to 5.0 Å, and the top 50% of NTMA+ to Sicube between 3.5 to 6.5 Å, at two different directions: seen from the face of the cube (left) and tilted along the z- and y-axis (right). | ||
Closer visual inspection of the cubic octamer reveals that it is not symmetrical but slightly “squashed” (see Fig. S12, ESI†), with rectangular faces. In a simulation study of H8Si8O12 by Catlow et al.,37 it was demonstrated that the cubic octamer has two frequently occurring conformations: a crown arrangement or a nonplanar hexagonal arrangement. The latter is the more energetically favourable conformation. The conformation that we find in this study is similar to the favourable, nonplanar hexagonal arrangement described by Catlow et al.
Conclusion
In this work we show that the D4R partially fragments in aqueous solutions, even at Si:TMA+ ratios of 1:1. The most abundant fragmented species are the monomer, dimer, S3R, S4R and D3R. This is an unexpected result as the D4R can be synthesised in a methanol–water mixture at a Si to TMA+ ratio of 1:1.6 The stability of the D4R is dependent on the overall Si concentration, and only at higher concentrations do the D4Rs remain intact in the solution. TAA+ ions, as well as short chain alcohols, have a stabilising effect on the D4R, with ethanol being more efficient than methanol. Sodium ions on the other hand demonstrate no stabilising effect. The D4R reveals a complex behaviour where standard additions to a silica materials synthesis can have both stabilising and destabilising effects, but the D4R also shows a predilection for associating with amphiphilic molecules.
The modelling results of the neutron scattering data reveal that TMA+ associate to the faces of the D4R. However, not all faces need to be shielded to provide stability. All silica species are anionic species, based on the orientation of water around these species, which is expected, as they are deprotonated. TMA+ strongly associates to the monomer, with a strong preference for the protonated sites. The monomer has thus an unshielded site, the deprotonated site – a site where, with time, polymerisation could occur.40
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to thank the ISIS Pulsed Neutron and Muon Source for allocation of experimental beamtime on NIMROD experiment number RB1210091. EJN and VA also acknowledge financial support from the Swedish Research Council via project grants 621-2011-4336 and 2015-05042, and the Linneaus grant Organising Molecular Matter (OMM) 239-2009-6794. KJE acknowledges funding from Lund University and the OMM grant for her visiting position.Notes and references
- D. B. Cordes, P. D. Lickiss and F. Rataboul, Chem. Rev., 2010, 110, 2081–2173 CrossRef CAS PubMed.
- R. Y. Kannan, H. J. Salacinski, P. E. Butler and A. M. Seifalian, Acc. Chem. Res., 2005, 38, 879–884 CrossRef CAS PubMed.
- S. Bourbigot, T. Turf, S. Bellayer and S. Duquesne, Polym. Degrad. Stab., 2009, 94, 1230–1237 CrossRef CAS.
- R. Q. Su, T. E. Muller, J. Prochazka and J. A. Lercher, Adv. Mater., 2002, 14, 1369–1373 CrossRef CAS.
- T. B. Reed and D. W. Breck, J. Am. Chem. Soc., 1956, 78, 5972–5977 CrossRef CAS.
- I. Hasegawa, S. Sakka, K. Kuroda and C. Kato, J. Mol. Liq., 1987, 34, 307–315 CrossRef CAS.
- I. Hasegawa, S. Sakka, K. Kuroda and C. Kato, Bulletin of the Institute for Chemical Research, Kyoto University, 1987, vol. 65, p. 5 Search PubMed.
- I. Hasegawa, S. Sakka, Y. Sugahara, K. Kuroda and C. Kato, J. Chem. Soc., Chem. Commun., 1989, 208–210 RSC.
- M. Wiebcke, M. Grube, H. Koller, G. Engelhardt and J. Felsche, Microporous Mater., 1993, 2, 55–63 CrossRef CAS.
- M. Haouas, D. P. Petry, M. W. Anderson and F. Taulelle, J. Phys. Chem. C, 2009, 113, 10838–10841 CrossRef CAS.
- D. P. Petry, M. Haouas, S. C. C. Wong, A. Aerts, C. E. A. Kirschhock, J. A. Martens, S. J. Gaskell, M. W. Anderson and F. Taulelle, J. Phys. Chem. C, 2009, 113, 20827–20836 CrossRef CAS.
- R. K. Harris and C. T. G. Knight, J. Mol. Struct., 1982, 78, 273–278 CrossRef CAS.
- R. K. Harris and C. T. G. Knight, J. Chem. Soc., Faraday Trans. 1, 1983, 79, 1525–1538 RSC.
- S. D. Kinrade, C. T. G. Knight, D. L. Pole and R. T. Syvitski, Inorg. Chem., 1998, 37, 4272–4277 CrossRef CAS PubMed.
- S. D. Kinrade, C. T. G. Knight, D. L. Pole and R. T. Syvitski, Inorg. Chem., 1998, 37, 4278–4283 CrossRef CAS PubMed.
- C. T. G. Knight, R. J. Balec and S. D. Kinrade, Angew. Chem., Int. Ed., 2007, 46, 8148–8152 CrossRef CAS PubMed.
- C. J. Brinker and G. W. Scherer, Sol-gel science: the physics and chemistry of sol-gel processing, Academic, cop., Boston, 1990 Search PubMed.
- J. S. Falcone, J. L. Bass, P. H. Krumrine, K. Brensinger and E. R. Schenk, J. Phys. Chem. A, 2010, 114, 2438–2446 CrossRef CAS PubMed.
- M. Haouas and F. Taulelle, J. Phys. Chem. B, 2006, 110, 3007–3014 CrossRef CAS PubMed.
- W. M. Hendricks, A. T. Bell and C. J. Radke, J. Phys. Chem., 1991, 95, 9513–9518 CrossRef CAS.
- K. T. Butler and D. W. Lewis, J. Phys. Chem. A, 2012, 116, 8786–8791 CrossRef CAS PubMed.
- S. Caratzoulas, D. G. Vlachos and M. Tsapatsis, J. Phys. Chem. B, 2005, 109, 10429–10434 CrossRef CAS PubMed.
- T. C. Ionescu, F. Qi, C. McCabe, A. Striolo, J. Kieffer and P. T. Cummings, J. Phys. Chem. B, 2006, 110, 2502–2510 CrossRef CAS PubMed.
- C. L. Schaffer and K. T. Thomson, J. Phys. Chem. C, 2008, 112, 12653–12662 CrossRef CAS.
- M. Kwasny, A. Kowalewska, K. Wodnicka and M. Handke, J. Mater. Sci., 2013, 48, 5188–5195 CrossRef CAS.
- K. S. W. Sing, Pure Appl. Chem., 1985, 57, 603–619 CAS.
- C. T. Kresge, M. E. Leonowicz, W. J. Roth, J. C. Vartuli and J. S. Beck, Nature, 1992, 359, 710–712 CrossRef CAS.
- T. Kjellman, N. Reichhardt, M. Sakeye, J.-H. Smått, M. Lindén and V. Alfredsson, Chem. Mater., 2013, 25, 1989–1997 CrossRef CAS.
- R. Ryoo and S. Jun, J. Phys. Chem. B, 1997, 101, 317–320 CrossRef CAS.
- A. K. Soper, Chem. Phys., 1996, 202, 295–306 CrossRef CAS.
- A. K. Soper, Mol. Phys., 2001, 99, 1503–1516 CrossRef CAS.
- A. K. Soper, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 72, 104204 CrossRef.
- A. K. Soper, J. Phys.: Condens. Matter, 2007, 19, 335206 CrossRef CAS PubMed.
- L. Liu, Y. Hu, L. Song, S. Nazare, S. He and R. Hull, J. Mater. Sci., 2007, 42, 4325–4333 CrossRef CAS.
- D. T. Bowron, A. K. Soper, K. Jones, S. Ansell, S. Birch, J. Norris, L. Perrott, D. Riedel, N. J. Rhodes, S. R. Wakefield, A. Botti, M.-A. Ricci, F. Grazzi and M. Zoppi, Rev. Sci. Instrum., 2010, 81, 033905 CrossRef CAS PubMed.
- A. K. Soper, GudrunN and GudrunX: programs for correcting raw neutron and X-ray diffraction data to differential scattering cross section, Oxfordshire, UK, 2011 Search PubMed.
- C. R. A. Catlow, D. S. Coombes, D. W. Lewis and J. C. G. Pereira, Chem. Mater., 1998, 10, 3249–3265 CrossRef CAS.
- E. J. Nilsson, V. Alfredsson, D. T. Bowron and K. J. Edler, Phys. Chem. Chem. Phys., 2016, 18, 11193–11201 RSC.
- C. J. Brinker and G. W. Scherer, Sol-Gel science: the physics and chemistry of sol-gel processing, Academic Press, 1990 Search PubMed.
- R. K. Iler, The Chemistry of Silica: Solubility, Polymerization, Colloid and Surface Properties and Biochemistry of Silica, John Wiley and Sons, Inc., 1979 Search PubMed.
- S. Sjöberg, Y. Hägglund, A. Nordin and N. Ingri, Mar. Chem., 1983, 13, 35–44 CrossRef.
- S. D. Kinrade and T. W. Swaddle, Inorg. Chem., 1988, 27, 4253–4259 CrossRef CAS.
- C. E. A. Kirschhock, R. Ravishankar, F. Verspeurt, P. J. Grobet, P. A. Jacobs and J. A. Martens, J. Phys. Chem. B, 1999, 103, 4965–4971 CrossRef CAS.
- C. T. G. Knight, J. Wang and S. D. Kinrade, Phys. Chem. Chem. Phys., 2006, 8, 3099–3103 RSC.
- J. Ambati and S. E. Rankin, J. Phys. Chem. A, 2010, 114, 12613–12621 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8cp05682j |
| This journal is © the Owner Societies 2019 |