
The Gigahertz and Terahertz spectrum of monodeutero-oxirane (c-C2H3DO)†

Abstract
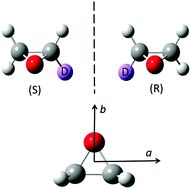
The rotational spectrum of monodeutero-oxirane was analysed as measured using the Zurich Gigahertz (GHz) spectrometer and our highest resolution Fourier Transform Infrared (FTIR) spectrometer system coupled to synchrotron radiation at the Swiss Light Source (SLS). 112 distinct line frequencies have been newly assigned in the GHz range (extended to 120 GHz, compared to previous work extending to only 59 GHz) including rotational states up to J = 23. We have furthermore assigned 398 lines in the far infrared or Terahertz range (0.75–2.10 THz or 25–70 cm−1) including transitions with rotational quantum numbers up to J = 59. The results are discussed in relation to the possible first astrophysical observation of an isotopically chiral molecule and in relation to molecular parity violation.
- This article is part of the themed collection: Challenges in spectroscopy: accuracy vs interpretation from isolated molecules to condensed phases
 Please wait while we load your content...
Please wait while we load your content...

