
Stabilization of two-dimensional penta-silicene for flexible lithium-ion battery anodes via surface chemistry reconfiguration†
 a
Houyang
Chen
*c
a
Houyang
Chen
*c
Abstract
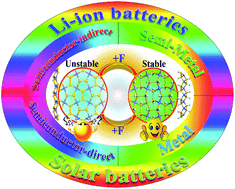
Silicon-based two-dimensional (2D) materials have unique properties and extraordinary engineering applications. However, penta-silicene is unstable. Herein, by employing first-principles calculations, we provide a facile surface chemistry method, i.e. functionalization, to acquire and reconfigure stable penta-silicene for use in flexible lithium-ion batteries. Our results of density functional theory calculations showed that the reconfigured penta-silicene nanosheets possess a broad range of properties, including semiconductors with an indirect bandgap, semiconductors with a direct bandgap, semimetals and metals. For fluorinated penta-silicene, a fluorine-concentration-induced transition from a semiconductor to a metal is found. For fully fluorinated penta-silicene, a mechanically induced transition from a semiconductor with an indirect bandgap to a semiconductor with a direct bandgap is obtained. Our calculation results showed the reconfigured penta-silicene is a high-performance anode for use in flexible lithium (Li)-ion batteries. A transition from a semiconductor to a metal with adsorption of Li atoms indicates a high electrical conductivity. It possesses low Li diffusion barriers (0.08–0.28 eV), demonstrating a high mobility of Li ions. The metallic feature and low Li diffusion barriers reveal that it has an ultrafast charge/discharge rate. This work suggests that surface chemistry reconfiguration provides new stable materials with excellent mechanical properties and tunable electronic properties for their promising applications in flexible metal-ion batteries and solar batteries as well as nanoelectronics devices.
 Please wait while we load your content...
Please wait while we load your content...

