
Water vapor uptake into hygroscopic lithium bromide desiccant droplets: mechanisms of droplet growth and spreading
 *ab
*ab
Abstract
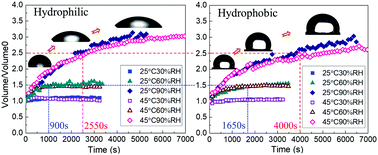
The study of vapor absorption into liquid desiccant droplets is of general relevance to a better understanding and description of vapor absorption phenomena occurring at the macroscale as well as for practical optimization of dehumidification and refrigeration processes. Hence, in the present work, we provide the first systematic experimental study on the fundamentals of vapor absorption into liquid desiccant at the droplet scale, which initiates a novel avenue for the research of hygroscopic droplet growth. More specifically we address the behavior of lithium bromide–water droplets on hydrophobic PTFE and hydrophilic glass substrates under controlled ambient conditions. Driven by the vapor pressure difference between the ambient air and the droplet interface, desiccant droplets absorb water vapor and increase in volume. To provide further insights on the vapor absorption process, the evolution of the droplet profile is recorded using optical imaging and relevant profile characteristics are extracted. Results show that, even though the final expansion ratio of droplet volume is only a function of relative humidity, the dynamics of contact line and the absorption rate are found to differ greatly when comparing data with varying substrate wettability. Droplets on hydrophilic substrates show higher absorption kinetics and reach equilibrium with the ambient much faster than those on hydrophobic substrates. This is attributed to the absorption process being controlled by solute diffusion on the droplet side and to the shorter characteristic length for the solute diffusion on hydrophilic substrates. Moreover, the apparent droplet spreading process on hydrophilic substrates when compared to hydrophobic ones is explained based on a force balance analysis near the triple contact line, by the change of liquid–vapor surface tension due to the increase in water concentration, and assuming a development of a precursor film.
 Please wait while we load your content...
Please wait while we load your content...

