DOI:
10.1039/C8CP04340J
(Perspective)
Phys. Chem. Chem. Phys., 2019,
21, 2251-2270
Decomposition of molecular properties
Received
10th July 2018
, Accepted 2nd January 2019
First published on 7th January 2019
Abstract
We review recent work on property decomposition techniques using quantum chemical methods and discuss some topical applications in terms of quantum mechanics-molecular mechanics calculations and the constructing of properties of large molecules and clusters. Starting out from the so-called LoProp decomposition scheme [Gagliardi et al., J. Chem. Phys., 2004, 121, 4994] for extracting atomic and inter-atomic contributions to molecular properties we show how this method can be generalized to localized frequency-dependent polarizabilities, to localized hyperpolarizabilities and to localized dispersion coefficients. Some applications of the generalized decomposition technique are reviewed – calculations of frequency-dependent polarizabilities, Rayleigh scattering of large clusters, and calculations of hyperpolarizabilities of proteins.
1 Introduction
The materials modelling community has during recent years increasingly turned its attention to multi-scale modelling. As time has passed, the replacement of the idea of one theory covering everything by the concept of bridging theories (or models) that each was limited to a certain scale in space and time, has become progressively more accepted. The most important variant of contemporary multi-scale modelling is given by the combination of quantum mechanics and classical physics, which in a sense gives the possibility to join the accuracy and rigour of the former with the applicability of the latter and which gives a possibility to find working approaches that efficiently can address the nanoscale. Thus, the joining of quantum mechanics (QM) with molecular mechanics (MM) with an expedient classical force field description of atoms has become a most important and popular area in contemporary in silico research with a wide variety of applied research areas. In the QM/MM approach the major types of intermolecular interactions are in principle accounted for both within and in-between the shells; electrostatic, polarization, van der Waals and short range (often though a QM/MM potential) and are mostly atomically decomposed. More recent development has concerned, in addition to structure and dynamics, also the “3rd dimension”, namely general properties, thus including optical properties, and also the development of QM/MM programs including multi QM cores in homogeneous and heterogeneous environments. Applications cover various types of spectroscopy and linear and non-linear properties of molecules in solution, on surfaces, in confined biological environments or in combinations of such environments. A key formulation of the response theory in a QM/MM setting is the work by Nielsen et al.,1 which has been diversified to various linear and non-linear electric and magnetic properties,2,3 to QM/MM/PCM,4 where a third layer constituting a dielectric continuum is added, and to resonant convergent response property formulations.5 The QM/MM response property theory has been implemented for different types of QM cores, like multi-configurational self-consistent field (MCSCF), coupled cluster and DFT. The various QM/MM methods can also be distinguished from one another by the way in which they describe the coupling between the QM and MM regions. The mainstream implementations are of DFT/MM type where originally the DFT to MM coupling has been limited to the description of the electrostatic interaction between the electron density of the QM region and point charges of the MM region.6 A more sophisticated description of the interaction between the MM and QM regions is achieved in the so-called effective fragment potential (EFP) method in which the coupling between the QM and MM regions includes electrostatic, polarization/induction, exchange repulsion, and a charge transfer interaction terms.7–9 For general response properties, the more recent research has focused on the DFT/MM response formalism which is capable of computing arbitrary molecular properties expressed via linear, quadratic and cubic response functions and their residues,10 and which includes full electrostatic, polarization/induction and dispersion terms in the description of the ground state interaction between a solute and its environment and the polarization/induction interaction in the generation of the response functions.1 It allows to consider a ladder of sophistication for the coupling between the QM and MM regions, referring to truncating the MM molecule or fragment electrostatic potential at different orders of the multipole expansion11 and by selecting different forms of the polarizability tensor for the MM molecule or the fragment.12–14 Various extensions from this outset have been made, for instance, to spin Hamiltonian parameters and paramagnetic open shell system,2,15 allowing for systematic studies of, electronic g tensors and hyperfine coupling constants of various radicals and spin labels.15 One can here also mention the recent extension of the QM/MM capacitance–polarization force field for embedding metal surfaces and/or nanoparticles in the metallic MM region in complex environments16—the so-called quantum mechanics/capacitance molecular mechanics (QM/CMM) model which provides a practical approach for theoretical modelling of properties of physisorbed chromophores on metal surfaces or nanoparticles. Here, the heterogeneous MM part is split into metallic and non-metallic parts, assuming a capacitance–polarization model for the electrostatics and polarization of the metallic part and distributed charges for the non-metallic part.17,18 The implementation of a heterogeneous MM region captures the essential physical features of the composite containing metal surfaces and/or nanoparticles and physisorbed chromophores in vacuum or solutions.
Force fields, either used in the preceding classical MD simulation or in the MM part of QM/MM, constitute crucial quantities as they dictate the precision and the cost, and therefore the limitations and applicability of the QM/MM calculations. It follows that force fields have been the subject of ever ongoing efforts of refinement and analysis where both the results of more accurate models, i.e. quantum mechanics based, and of fitting to experimental data are employed. Largely, such force fields divide into electrostatic, polarizable and reactive force fields regulating charge transfer in the molecular mechanics medium. With the implementation of QM/MM models the quality of force fields are also scrutinized in terms of the interaction between the quantum and classical parts. One has during recent time witnessed new techniques to generate force fields for QM/MM calculations and techniques for integrating dynamics into the property calculations through QM/MM—the so-called integrated approach.19 An ultimate aim is to use the same QM/MM parameterization for the dynamics as for the property calculations, a next level in ambition is to use the same force field parameterization in classical molecular dynamics as the one used in the MM part of the QM/MM property calculation. None of these two ambitions, are, however, fulfilled in common calculations published in literature. In some sense the requirement of the MM force field in QM/MM is higher than for the force field in pure MD, especially the paramount role of polarization and the need for a fine granulation of the force field. Thus even for the water molecule there is need to granulate the charges of the molecule as well as to granulate the polarizabilities. This puts additional demands in that the quality of the force fields that should balance the precision of the quantum part for the evaluation of the structural dynamics or properties. More recent implementations of properties and spectroscopy in the QM/MM framework often demand full granulation of such embeddings in terms of separate atomic contributions, sometimes even covering atom–bond partitioning and multipole expansions of the charges. An often attended vehicle to fulfil this demand is the LoProp method20 for extracting atomic and interatomic contributions to molecular properties. It was originally formulated as a sequence of transformations of the atomic overlap matrix, giving a localized orthonormal basis that depends only on molecular structure and the initial atomic orbital basis set. The localized properties are then obtained by forming partial traces over basis functions associated with one chosen centre or a pair of centres. Thus by means of LoProp several systematically connected models for the solvent interaction can be formulated, ranging from a simple molecular point charge to bond- and atom-centred multipoles and polarizabilities. The precision needed, and thus the level of decomposition, is to a large extent dictated by the property that is to be calculated. This level also has a bearing on the standard problem how to split the QM and MM parts of the system in a QM/MM calculation, for instance, the need to bring in water molecules in the QM part is often relieved by a careful LoProp MM parameterization of water.3 To the known limitations of the LoProp method one can mention the tendency to “overpolarize” the interaction, especially to the QM part close to the QM/MM interface but also between closely encountering MM molecules. The (ad hoc) introduction of distributed point charges and polarizabilities by Gaussian broadenings remedies the problem to some extent.16 These localized properties have indeed been of much use as granulated force fields in QM/MM calculations of properties, ranging from NMR, EPR over to optical properties and X-ray spectroscopy.1,5,21–24
In a previous work25 we showed how a LoProp polarizable force field can be derived quantum mechanically for extended environments by using analytical response theory. The theory was recently extended to construct the localized polarizable force fields to be frequency dependent.26 The possibility to do so follows from the fact that analytic response theory can determine the first-order response for frequencies which directly match those of the externally perturbing field. This effectively produces polarizable force fields which include the quantum mechanical density perturbation caused by an external field. This extension of the MM region in typical QM/MM calculations allows for a more physical description of properties in the QM region, since the interaction between the MM and the QM regions now includes the frequency dependence of the classical region. Further development in terms of non-linear hyperpolarizable force fields followed, which, e.g. can be used to reconstruct hyperpolarizable tensors, and properties derived therefrom, in large molecular clusters and proteins.27 Further recent development has concerned the decomposition of obtain complex frequency dependent polarizabilities and dispersion coefficients. The centring of such polarizabilities to bonds is also possible, and, more importantly, to higher order dispersion coefficients, higher than C6, is doable if the underlying property integrals are at hand. It could allow the development of chemically dependent local dispersion interaction force fields to be introduced in molecular dynamics. In addition to the use in QM/MM, the LoProp technique has recently been shown to be powerful tool for building up “classical” properties of large molecular clusters, like Rayleigh scattering of atmospheric nanoparticles,28 and hyperpolarizabilities (second harmonic generation) of proteins.29 In the present work review recent development of force field decomposition and local property calculations with quantum chemical methods and discuss some topical applications both in terms of QM/MM and for building up properties of large molecules and clusters.
2 Theory
2.1 The LoProp transformation
Gagliardi et al.20 introduced the LoProp method for extracting atomic and interatomic contributions to molecular properties. It was formulated as a sequence of transformations of the atomic overlap matrix, giving a localized orthonormal basis that depends only on molecular structure and the initial atomic orbital basis set. The localized properties were obtained by forming partial traces over basis functions associated with one chosen center or a pair of centers. The aim was to obtain physically sound and transferable local properties which sum up to the molecular property and which are reasonably constant for a given atom in chemically similar systems. The implications of this is that these localized properties can be used as force fields in QM/MM calculations.
The requirement on the initial basis set is that the atomic orbitals can be grouped into occupied and virtual orbitals, e.g. if they resemble the actual orbitals in atomic states. Given this division, the sequence of transformations can be briefly summarized as: for the combined atomic-orbital basis set of the molecule, orthonormalize first within atomic subblocks, second within the combined occupied AO space. Then project out any occupied components in the virtual AO space, and finally orthonormalize the virtual space. This leads to an orthonormal localized basis set, the LoProp basis, where each orbital is associated with an atomic center.
These steps generate a total matrix Tμl describing a transformation between the AO:s (ξμ) and the LoProp (ϕl) basis functions
| |  | (1) |
For integral representations of one-electron operators
| |  | (2) |
we thus have the transformation rule
| | 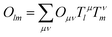 | (3) |
while for the density matrix we have the inverse transformation
| |  | (4) |
Next the atom and “bond” contributions are given by grouping the summations involved in the trace operations into partial traces over the atomic subsets, denoted by A,B…
| |  | (5) |
The localized charges and isotropic polarizabilities are the parameters which form the force fields for the QM/MM calculations in this work.
The decomposition of the total electronic charge according to eqn (5) gives
| | 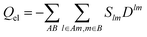 | (6) |
where
S is the overlap matrix. As the LoProp basis is orthonormal (
Slm =
δlm) as well as localized this becomes
| |  | (7) |
i.e. there are no bond contributions to the total charge and the localized charge associated with an atomic site
A with nuclear charge
ZA, is
| |  | (8) |
2.2 Analytical local polarizabilities
We present here an alternative formulation and implementation of the LoProp approach. The original implementation was based on finite field perturbation theory applied to localized dipole moments. Here we apply analytical response theory where the change of a wave function in an external field is represented by a unitary transformation. The theory derives from the classical work by Olsen and Jørgensen30 for MCSCF theory and later generalized for DFT by Sałek et al.31 Following the notation of the latter, we have that a wave function |0〉 is transformed by a unitary operator as a result of the perturbation| | |![[0 with combining tilde]](https://www.rsc.org/images/entities/char_0030_0303.gif) 〉 ≡ e− 〉 ≡ e−![[small kappa, Greek, circumflex]](https://www.rsc.org/images/entities/i_char_e109.gif) |0〉 |0〉 | (9) |
The operator κ has a real anti-symmetric matrix representation which in second quantization is given by| | | = κpqa†paq | (10) |
If we restrict to static perturbations, the Ehrenfest theorem gives that the first-order properties can be derived from| | δ〈0|[![[Q with combining circumflex]](https://www.rsc.org/images/entities/i_char_0051_0302.gif) ,eκHe−κ]|0〉 = 0 ,eκHe−κ]|0〉 = 0 | (11) |
for an arbitrary operator . By choosing the same excitation operators that are used to expand we obtain a system of equations for the unknowns, the matrix elements κpq. As the details of these derivations are quite lengthy we refer the interested reader to ref. 31.
After solving the linear response eqn (11), some matrix algebra allows us to express the first-order change of an expectation value in terms of a trace of the product of the operator matrix and the first-order density matrix
| | 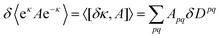 | (12) |
where the first-order change in the density matrix can be expressed as
all in MO basis. It is now straightforward to transform the same quantities to AO (with MO-coefficients
Cμp) or LoProp basis (with LoProp transformation
Tμl)
| |  | (14) |
With the first-order densities at hand we are now in a position to proceed to evaluate the localized polarizabilities in the LoProp approach. These were introduced in ref. 20 by considering the electric field derivatives of the molecular dipole moment with respect to an arbitrary gauge origin C
| | 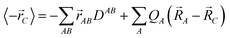 | (15) |
![[r with combining right harpoon above (vector)]](https://www.rsc.org/images/entities/i_char_0072_20d1.gif) AB
AB is the electronic coordinate with respect to the midpoint between atoms
A and
B, if
A ≠
B, or with respect to an atomic site
![[R with combining right harpoon above (vector)]](https://www.rsc.org/images/entities/i_char_0052_20d1.gif) A
A if
A =
B. Considering at first a first-order variation of the dipole moment
| | 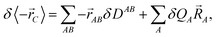 | (16) |
it is seen that the last term in
(15) containing
C vanishes due to charge conservation. The fact that the individual terms in
(16) are origin dependent were solved by Gagliardi by introducing charge transfer between atomic sites – an anti-symmetric charge transfer matrix Δ
QAB satisfying
| |  | (17) |
where the charge shift on a site
A is
| |  | (18) |
This leads to a localized polarizability of the form
| | | αAB = δ〈−〉AB = −ABδDAB + ΔQAB(A − B) | (19) |
The charge transfer matrix is not uniquely defined but it solves the problem with gauge-dependence and one gets a physically reasonable interpretation of the local polarizability – as a change of the local dipole moment and, in the case of bond contributions, a charge shift between the two sites
A and
B. In our calculations we redistribute the bond contributions to their atomic sites and only consider local atomic polarizabilities.
To summarize, the main difference between this approach and the original implementation is that it is based on calculations on analytical response theory rather than finite-field perturbation theory. An advantage of the formulation is that it is straightforward to generalize the LoProp concept to local dynamical polarizabilities and to higher order polarizabilities. The analytical LoProp code is a post-processing tool for Dalton32 written in the Python programming language.
2.3 A frequency-dependent LoProp approach
In a previous paper25 we demonstrated an alternative derivation of the LoProp approach which was based on analytical response theory. As response theory provides a formulation of perturbation theory which handles time-independent and time-dependent cases within the same formalism, we then investigated the effects of projecting the dynamical polarizability onto atomic contributions. This can be expected to be meaningful provided that the frequencies involved are well below the first resonance. To outline the steps in single-determinant time-dependent response theory (see e.g. Salek et al.33) we have a state |〉 that evolves in time| | | |〉 ≡ e−(t)|0〉 | (20) |
where the exponential operator is a parameterized time-evolution operator, a real anti-hermitian operator| | | = κpq(t)a†paq | (21) |
of which the matrix elements {κpq} form the parameters of the theory. A time-dependent variational principle based on the Ehrenfest theorem provides a solution to the time-independent Schrödinger equation in this space of parameters {κpq}, i.e. arbitrary static operators Ô that satisfy| |  | (22) |
A well-chosen set of operators {Ô} provides linear systems of equations for the parameters {κpq} to various orders in the perturbation. If we consider monochromatic perturbations of frequency ω we can extract the dynamical dipole polarizability from the linear response function| |  | (23) |
| | | δDpq(ω) = [δκT(ω),D]pq | (24) |
The dipole moment is expanded in terms of atomic- and bond contributions
| | 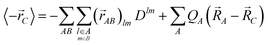 | (25) |
with the localized LoProp basis, where
AB is the electronic coordinate with respect to the midpoint of the bond
A–
B and the frequency-dependent variation can be written
| |  | (26) |
where Δ
QAB is a charge transfer matrix satisfying
| | 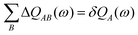 | (27) |
and where the local charge response to the external perturbation is
| |  | (28) |
This leads to a localized dynamical polarizability of the form
| | | αAB(ω) = δ〈−〉AB = −ABδDAB(ω) + ΔQAB(ω)(A − B) | (29) |
completely analogous to the dynamic case.
Most applications of classical models for the calculation of polarizabilities and hyperpolarizabilities are semi-empirical in nature, e.g. in the work of Applequist et al. the atomic polarizabilities are parameters which are optimized for a test set of molecules. The approach reviewed above, on the other hand, is fully ab initio in nature in that quantum mechanical values for a monomer are used in the classical models for a cluster of molecules. For clusters of moderate size the validity of the model can be tested with a full quantum mechanical calculation for smaller oligomers. It can be expected that these models hold for well separated systems, while breaking down at close distances where the exchange interaction between the overlapping electron densities is non-negligible. After highlighting the basic point dipole models, we derive the equations necessary for the extraction of atomic hyperpolarizabilities.
2.4 Point dipole models
2.4.2 Extension: polarizability of a system of hyperpolarizable particles.
Now we extend the Silberstein–Applequist model to a system of particles with additional structure, with local polarizabilities as well as hyperpolarizabilities. With a local hyperpolarizability tensor βi the local dipole moments are| | 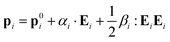 | (41) |
and consequently the induced components are given by| | | δpi = αi·δEi + βi:EiδEi | (42) |
The first question displays how this affects the definition of the total polarizability α
| |  | (43) |
As before, collecting induced dipoles to the left we obtain
| |  | (44) |
We now note that the structure of this equation is the same as eqn (35) with a corrected local polarizability
| | ![[small alpha, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0dc.gif) i = αi + βi·Ei i = αi + βi·Ei | (45) |
This equation is though different from
eqn (35) in the sense that it refers to the explicit value of the local electric field at each site. As we are considering the effect of an external electric field on the total system we assume these local fields to be the stationary value in the absence of an external field,
i.e.| |  | (46) |
Nevertheless, it is now not sufficient to invert a matrix – we also have to solve for the local fields iteratively in order to set up the linear systems of equations for the dipole shifts. This gives an Applequist equation with corrected local polarizabilities
| |  | (47) |
or
| | 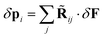 | (48) |
where
| | ![[R with combining tilde]](https://www.rsc.org/images/entities/char_0052_0303.gif) ij = (1δij − i·Tij)−1·j ij = (1δij − i·Tij)−1·j | (49) |
2.5 Hyperpolarizability of a system of hyperpolarizable particles
The next question displays how an equivalent model can be defined for at total hyperpolarizability. Taking the same argument from the beginning and considering the change in the individual dipole moments we obtain| |  | (50) |
Now we are only looking for terms quadratic in the first-order field so we can set δ2F = 0. The second-order effect in the local field is| |  | (51) |
giving| | 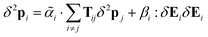 | (52) |
The equation for the second-order dipole moments are thus| | | (δij1 − iTij)·δ2pj = βi:δEiδEi | (53) |
From the linear problem we have| |  | (54) |
which in the case of a uniform external field simplifies to| | 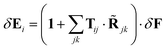 | (55) |
Analogously to the Applequist relations, the molecular hyperpolarizability is now the sum of local second-order dipole shifts
| |  | (56) |
2.6 LoProp hyperpolarizability
In a previous section we showed how the localized dynamic polarizabilities of atoms can be extracted by analytical response theory at static or dynamic frequencies for a molecule.26 We now extend the formulation to include the contribution of a higher-order perturbation to the electronic density, to obtain localized hyperpolarizabilities.
As for the LoProp frequency dependent polarizability, see Section 2.3, we start out from response theory for single-determinant wave-functions,31 where the time-dependence of an electronic state can be written as the time-evolution operator acting on a stationary state
| | | |〉 ≡ e−κ(t)|0〉 | (57) |
where the exponential operator is a time-evolution operator and the operator
κ(
t) is the anti-hermitian operator with matrix elements
The elements of κ(t) forms the parameters of the theory. Furthermore, a time-dependent variational theory based on the Ehrenfest theorem can be expanded to give a solution to the time-independent Schrödinger equation in terms of the parameters κ(t)pq
| |  | (60) |
where the operator
Ô can be any time-independent property operator. Choosing
Ô as the non-redundant orbital-excitations gives a linear system of equations that determines the parameters
κrs uniquely.
30 Choosing
Ô as well as the perturbation
V to be dipole moment operators
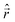
, (the latter corresponding to the dipole interaction between the molecule and an external electric field with frequency
ω) we can extract the polarizability as the linear response function
| | 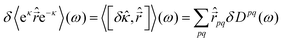 | (61) |
| | | δDpq(ω) = [δκT(ω),δD]pq | (62) |
and the hyperpolarizability as the quadratic response function
| |  | (63) |
where
| |  | (64) |
To obtain localized properties, the LoProp transformation is then applied to yield a localized orthonormal basis, where the molecular dipole moment can be written as the sum of atom and bond contributions
where
AB is the electronic coordinate with respect to the bond-center between the atoms
A and
B. The first two terms are the contributions from the polarizability, and the last two terms are from the hyperpolarizability. Δ
QAB(
ω) and Δ
2QAB(
ω) are the atomic charge-transfer matrices, both individually satisfying
| | 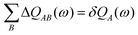 | (65) |
| | 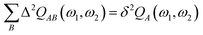 | (66) |
where
| |  | (67) |
| |  | (68) |
are the first, and second-order local response with respect to the external field. Having the second-order electronic density perturbation, the final form of the localized hyperpolarizabilities reads
| |  | (69) |
We emphasize that in the original work static hyperpolarizabilities (
ω1 =
ω2 = 0) were considered. These relations have been implemented as a plugin for Dalton
32 available in the public domain.
49
2.7 Dispersion interaction
We here summarize the relations between the linear response functions and dispersion energies. The detailed derivations are based on the work by Magnasco et al.50 and are available in ref. 51.
Given the density operator in second quantization, the density–density response function of a molecular system can be written as
| | 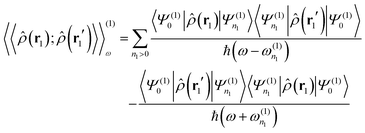 | (70) |
It is well established that the dispersion energy can be expressed as an integral over polarizabilities over imaginary frequencies. This gives
| |  | (71) |
A Taylor expansion with respect to intermolecular distance yield to lowest order an interaction that decays as
R−6| |  | (72) |
where the dipolar coupling tensor is
| | 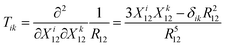 | (73) |
The isotropic expression is obtained by a rotational averaging procedure52
| |  | (74) |
which involves orientation averaging of coupling coefficients between body-fix and space-fix coordinate axes:
| | 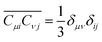 | (75) |
Substituting with the isotropic polarizabilities
| |  | (76) |
we obtain the final expression
| |  | (77) |
The LoProp approach20 to obtain frequency dependent polarizabilities is based on diagonalizing the atomic overlap matrix S. This is done as described in Section 2.1 in sequential orthonormalization steps which gives a transformation matrix used to transform the property integrals and perturbed electronic densities. By summing the transformed integrals over the subspace of atom-pairs, the properties in the LoProp basis are obtained. It was shown above how the frequency-dependent polarizability and hyperpolarizability could be obtained from linear response theory.26,27 Here, for the dispersion interaction, the natural extension to the extraction of the response vectors from the CPP code is made in order to obtain the distributed real polarizabilities of atoms at imaginary frequencies.
2.8 Damping
Due to very large molecular polarizabilities caused by the induced dipoles at short inter-atomic distances, the original Applequist34,37 equations were modified by Thole.53 The main difference in the Tholes model derives from the introduction of the so-called damping parameters, which depend on the scaled inter-atomic distances. The equation used to calculate the damping reads| |  | (78) |
which gives the following damping parameters for the potential, field, and field-gradient, respectively| |  | (79) |
| |  | (80) |
| |  | (81) |
where v = au, and a is an arbitrary damping parameter. The parameter derived most accurately using the largest set of data54 is a = 2.1304, and is the one also used in this review.
In the damped model, the dyadic tensor now reads
| |  | (82) |
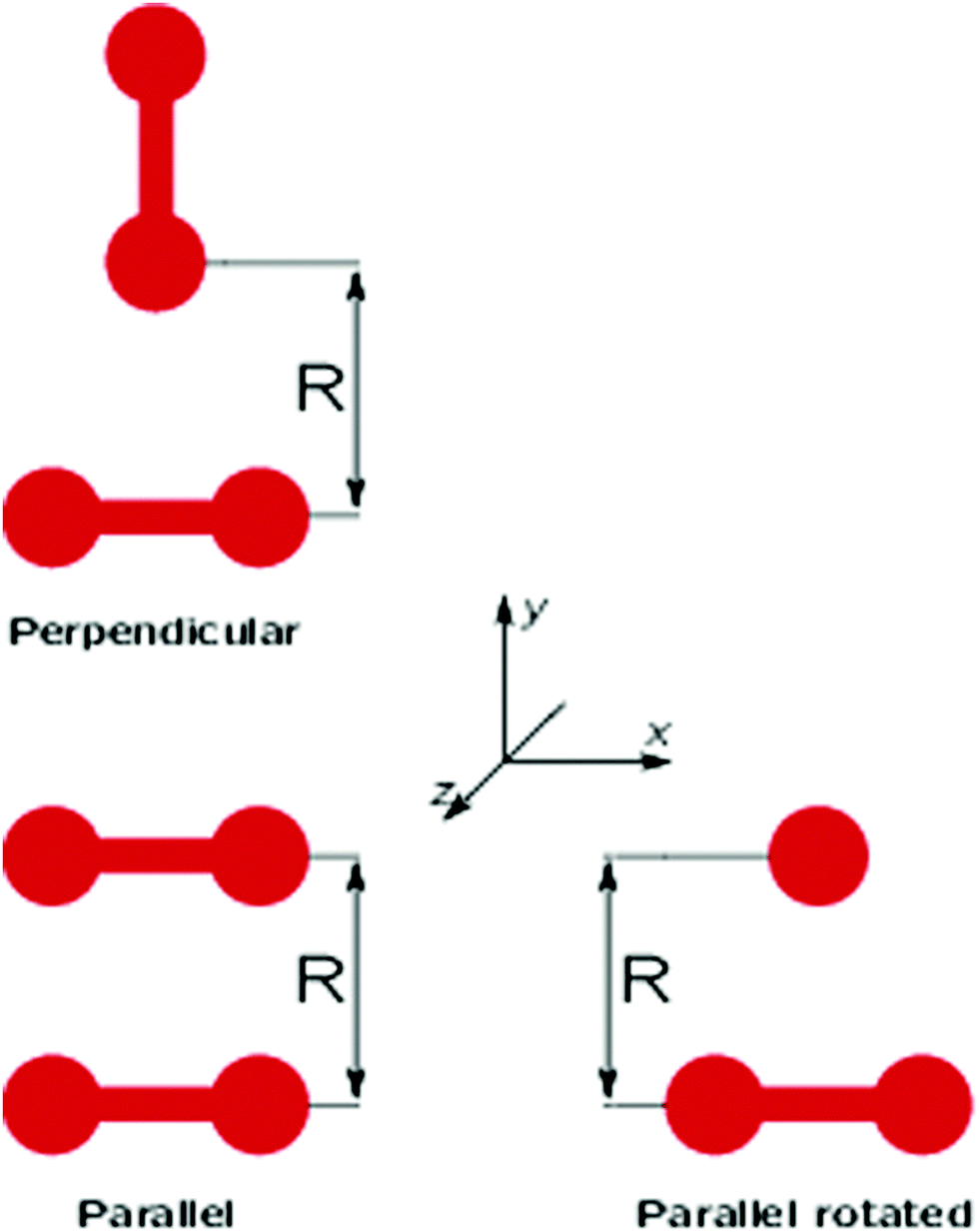
As an illustration of the damped model,
Fig. 1 shows the
Ex and
Ey electric-field components in the
xy-plane stemming from a position in origo, with (left column) and without (right column) damping.
 |
| | Fig. 1 The Ex and Ey components due to an ideal point charge at origo with q = 1 a.u. The right column illustrates how Tholes exponential damping gradually decreases the electric field near the charge source. From ref. 29. Copyright ACS Publishing. | |
3 Decomposition of frequency dependent polarizabilities
The polarizability is an example of a property for which both classical and quantum models have played important roles for its calculation. In the original works by Applequist, Carl, and Fung37 and Silberstein35,36 the concept of the polarizable point-dipole model was introduced in which the molecular polarizability is obtained as a result of atomic interactions governed by the localized polarizability tensors of each point-dipole (see Theory section for mathematical details). In that formulation, the induced dipoles are determined solely by the atomic polarizabilities which in turn can be directly calculated by solving a system of linear equations. The atomic polarizabilities were then often fitted to yield molecular polarizabilities as closely as possible to molecular polarizabilities obtained from experimental refractive index measurements.55 As shown already in the original works, short interatomic distances could give rise to polarization catastrophes since the induced dipoles then would increase indefinitely. Several models have subsequently been put forward to fix the problems arising from short interatomic distances, for example, Thole53 tested two modified dipole-interaction models, based on charge distributions, also called the linear- and exponential models, and re-parameterized the atomic polarizabilities to fit the new interaction equations: Ren and Ponder included higher-order multipole induction terms, in order to study the intra- and intermolecular interactions56 of molecules, with successful extensions to the polarizable atomic multipole water model.57 Wang et al.58,59 further extended the point-dipole model with new sets of experimental data available to molecules.60 Jensen et al.,39 and others, used quantum mechanical calculated molecular polarizabilities to fit atomic polarizabilities. Here a quantum approach was used to evaluate frequency dependent molecular polarizabilities of amino acids and proteins,40 and also later the second hyperpolarizability.41
Ref. 26 presented a way of incorporating quantum mechanical granulation of a molecular mechanics environment through the localization of frequency dependent polarizabilities. The work was motivated by that such frequency dependent molecular mechanics force fields allow for an instantaneous, optical, polarization in the molecular mechanics part by the electronic degrees of freedom in the quantum part of a quantum mechanics/molecular mechanics (QM/MM) model and make it possible to extend the QM/MM properties to cover the full QM/MM simulation volume. This is a generalization of the QM/MM technology where the embedding MM part acts as a perturber of the property determined by the QM. Furthermore, these force fields allow for calculations of frequency dependent properties for very large clusters that are evaluated classically. The construction of frequency dependent localized polarizable force fields within analytic response theory, makes it possible, in contrast to the finite field algorithm, to determine the first order response for frequencies which directly match those of the externally perturbing field. That includes the implementation of the modified Silberstein–Applequist mode, alluded to above, for many interacting induced dipoles for the linear frequency dependent polarizability. The performance and precision of the method was evaluated in ref. 26 through studying a few selected cases. The purely classical intermolecular polarizability using ab initio derived properties was also studied, and, specifically, the effect caused by frequency dependency in the MM environment in a QM/MM study of water clusters, and the effect of the dynamic local polarizability in a larger delocalized aromatic system.
The performance of the frequency dependent LoProp method was made in ref. 26 on the static and dynamic polarizability of the TIP3P water model and within a study of the size dependent behavior and convergence of the mean polarizability for different ensembles of water clusters. Two different cases were studied – the error in the mean polarizability using the modified Silberstein–Applequist model for water in gas phase was calculated by means of QM response theory, and the QM/MM response method was then used to obtain the absolute mean polarizability. That was done for the static, frequency independent case, and for frequency dispersion. Also the role of the actual decomposition scheme was explored, thus employing different water models for the polarizability, as well as the parameterization of the underlying response theory for the numerical outcome and convergence behavior of the polarizabilities. The LoProp scheme was used for construction of decomposed force fields with five main types typically used in QM/MM applications:61 (i) atom distributed charges; (ii) atom distributed charges with a central polarizability; (iii) atom distributed charges and polarizabilities; (iv) atom and bond distributed charges and polarizabilities; (v) atom and bond distributed charges and polarizabilities with multipoles expanded at each atomic site (normally truncating at octopoles). These different MM models have been particularly well tested in the case of the water solvent. The precision of the more refined models is associated with a computational cost, and is also directed by the type of property considered in the QM/MM calculations. It has been noted on several occasions that a good MM parameterization of water (and possibly any other solvent), relieves the need to include waters in the QM box, thereby making the QM/MM partitioning remain well-defined over simulation time, and substantially reducing cost.62
It was found in ref. 26 for water that the variation in the frequency dependent polarizabilities gives only a small perturbation of the QM wave function in QM/MM calculations and, indeed, that the distributed LoProp point dipoles outperform the molecule-centered ones by 10%. Furthermore, it was shown that the bulk frequency dependent α(ω) prediction is 25% better for properties obtained at the corresponding frequency in comparison with the properties obtained in the static limit.
QM/MM results for the water clusters are summarized in Fig. 2 and 3. It is seen that the effect of including frequency dependent polarizabilities in the MM region does not affect the ![[small alpha, Greek, macron]](https://www.rsc.org/images/entities/i_char_e0c2.gif) Cluster with any significance. If the amount of water molecules in the MM region is small, then the mean polarizability for one QM molecule is larger than for the gas phase, but as the MM region is systematically expanded, the mean polarizability approaches the gas-phase value for the oxygen-centered type, and only a slight underestimation of the gas-phase value is seen. For one water molecule in the QM region, the QM/MM interaction stagnates at about 30 molecules in the MM region, which shows that the QM/MM interaction is only sensitive to the most close-lying MM segments.
Cluster with any significance. If the amount of water molecules in the MM region is small, then the mean polarizability for one QM molecule is larger than for the gas phase, but as the MM region is systematically expanded, the mean polarizability approaches the gas-phase value for the oxygen-centered type, and only a slight underestimation of the gas-phase value is seen. For one water molecule in the QM region, the QM/MM interaction stagnates at about 30 molecules in the MM region, which shows that the QM/MM interaction is only sensitive to the most close-lying MM segments.
 |
| | Fig. 2
(ω) obtained by varying the size of the QM region (x-axis denotes number of QM waters) for in total 100 water molecules. From ref. 26. Copyright Royal Academy of Sciences. | |
 |
| | Fig. 3
(ω) obtained by varying the size of the MM region for one QM water molecule. From ref. 26. Copyright Royal Academy of Sciences. | |
As an example of a bonded polymer the frequency dependent mean polarizability for a tryptophan residue, embedded in an MM polarizable environment, was investigated in ref. 26. In Fig. 4 the frequency dependence of the isotropic αLoProp(ω) for all the atoms in tryptophan is summarized. The atom or group contribution was given to the total polarizability over a number of snapshots.
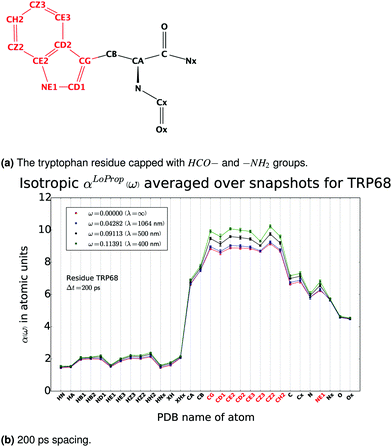 |
| | Fig. 4
α
LoProp(ω) averaged over 10 snapshots for the residue TRP68. From ref. 26. Copyright Royal Academy of Sciences. | |
For larger systems, and especially polarizable systems, the effect of frequency dependence in the MM environment is expected to become non-negligible, especially if the frequence close up to resonances. The decomposition can directly be implemented with any quantum mechanical wave function and basis set, an extension of the studies above for any kind of systems is in principle possible and the frequency independent force fields in QM/MM calculations can routinely be replaced by frequency dependent ones. In a recent paper by Norby et al.63 it was shown that the introduction of a frequency-dependent embedding potential leads to further model complications upon solving the central QM/MM equations defining specific molecular properties. It was also shown that from a numerical point of view that the consequences of using such a frequency-dependent embedding potential is in general small except in cases if the absorption bands of the environment are close to that of the region treated using quantum mechanics.
4 Decomposition of hyperpolarizabilities
The LoProp transformation of charges, multipole moments and multipole polarizabilities presented above, thus offers a way to go beyond numerical fitting of parameters to physical observables—that holds also for non-linear properties, including the first order hyperpolarizability. In the work of Harczuk et al.27 hyperpolarizabilities were obtained through combining the LoProp transformation and the second-order Applequist formalism, in which the point dipoles are additionally described by their non-linear polarization properties, i.e. also including the first hyperpolarizability to the original Applequist equations (for mathematical details see Theory section). The equations for the LoProp hyperpolarizabilities were extracted from the second order response of a molecule subjected to an external field using analytical response theory, where the higher order terms to the dipole moment induction thus were obtained from the non-linear contribution of the first hyperpolarizability. The theory builds on the afore mentioned original formulation of Silberstein, later developed by Applequist et al., where the interaction of point dipoles is described by point polarizabilities. The work presented in ref. 27 is the first known classical formulation of non-linear induction of point dipoles and is as such a natural extension of the earlier many-interacting polarizable point-dipoles models. As the formulation describes the molecular and cluster polarizability and hyperpolarizability of interacting molecules through their atomic site properties, it can so be used to evaluate macroscopic first order hyperpolarizabilities, something which is computationally expensive and most often unfeasible for large systems using purely ab initio methods.
For clusters of moderate size the validity of the model can be tested with a full quantum mechanical calculation for smaller oligomers. As for the corresponding polarizability, it can be expected that these models hold for well separated systems, while breaking down at close distances where the exchange interaction between the overlapping electron densities is non-negligible. In the work of Harczuk27 applications were made on water clusters because of its anomalously varying first order hyperpolarizability with respect to phase, originally observed by Ward and Miller64 and by Levine and Bethea,65 and later calculated for the first solvation shell by Mikkelsen et al.66,67 The water dimer and pentamer systems and clusters up to 25 water molecules were used as a fist benchmark of the methodology.27 A large-scale calculation on a water cluster containing 500 water molecules was finally performed, which demonstrated that the proposed method can lead to opportunities in property predictions of large, previously unattainable, systems. At small intermolecular distances, the overlap between the electronic wave functions of the water molecules is non-negligible, and thus the classical model breaks down, failing to describe response properties in an accurate manner. However, at distances when the classical model is expected to give large errors, r < 2.5 Å, the oxygen in the water molecules, responsible for most of the polarization due to an external field, rarely encounter each other, which can be seen by the oxygen–oxygen radial distribution function obtained from X-ray diffraction68 of liquid water at room temperature and pressure. It may thus indicate that it is still valid to compute the optical response using a classical formalism for liquid water.
In Fig. 5, the relative error of the total dipole moment for two water molecules in the same plane at a variable separation is plotted versus intermolecular distance. The relative error in the total induced dipole moment, referring to properties situated at the centre of the water molecules, is seen to be largest for the simple model, while the LoProp model decreases the error significantly. With the resolution given in the figure, the two LoProp curves overlap and the two “Simple” curves overlap almost complete. It is notable that the asymptotic behaviour is the same for all models and approaches the QM result at large distance. The zeroth model, which does not include any interactions, perfectly aligns with the additive method, which is a summation of the molecular dipole moments as defined by the LoProp basis.
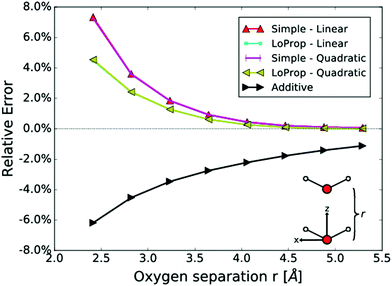 |
| | Fig. 5 The relative error of the total induced dipole moment for two water molecules as a function of the intermolecular distance. The additive method includes no polarization of the point-dipoles. From ref. 27. Copyright Royal Academy of Sciences. | |
In Fig. 6 the absolute value of the isotropic β‖ component, calculated with the TDHF/TDDFT methods, is plotted against the number of molecules in water clusters. The TDHF and the TDDFT methods give quite different results for the QM part, but share the characteristics that the value of β‖ decreases for N < 16 and then rapidly increases by a large magnitude. As the water cluster configuration is taken from a random MD snapshot, the properties will depend on the unique geometry, but the difference in QM methods is still quite noticeable. We see in Fig. 6 that the LoProp model agrees better with TDDFT using the B3LYP functional than with TDHF. The latter method greatly underestimates the first hyperpolarizability in conjugated systems, while TDDFT often yields too large hyperpolarizabilities due to its underestimation of the excitation energy levels. These trends are well known and refers in general to overestimation, respectively, underestimation of the HOMO–LUMO gaps. As the hyperpolarizability changes its sign for the small but non-zero βyyz component, it will affect the induced polarizabilities in eqn (45) differently from the TDHF. The reverse sign of the βyyz component for TDDFT could thus be an additional reason why TDDFT performs differently from TDHF in this case. Likewise, since all the polarizability and hyperpolarizability components increase in magnitude from TDHF to TDDFT, the quadratic Applequist model yields larger values of the local fields, leading to an increased hyperpolarizability β‖.
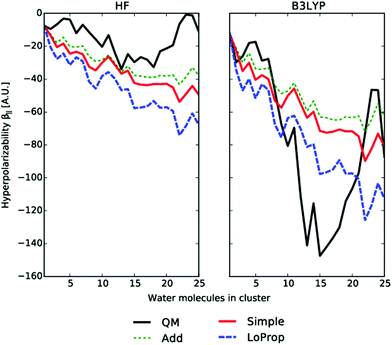 |
| | Fig. 6 The absolute value of the parallel component of the first hyperpolarizability calculated for clusters containing varying amounts of water molecules. The basis set used is the 2s (H)/4s3p1d (O) ANO type. From ref. 27. Copyright Royal Academy of Sciences. | |
In Fig. 7, results are shown for calculations of all properties using the quadratic Applequist formalism for a cluster consisting of 500 water molecules. It can be seen that the polarizability is not so heavily affected by the long-range interactions in this model, as it converges fairly quickly, dropping 2.9% from the case where only the closest lying neighbours affect each other. For the dipole moment, the situation is different, as including the interaction of more distant lying particles can change the value quite drastically. For the hyperpolarizability, the situation is similar to the dipole moment but with the difference that it oscillates more strongly in absolute value and even changes its sign at intermediate values of the cut-off screening. This phenomenon was previously confirmed by multi-configuration self-consistent reaction field theory,66,67 where the observed sign change was shown to occur for the water pentamer. In the quadratic Applequist method, this intermediate sign change occurs due to the lack of inclusion of long range fields for the inducible point dipoles. At full interaction, we see that all properties converge, and that the first hyperpolarizability takes a negative value.
 |
| | Fig. 7 Convergence of the isotropic analogues of the dipole moment, polarizability, and hyperpolarizability of a snapshot cluster with 500 water molecules. The x-axis represents the range of the field that the inducible point dipoles are affected by. Furthermost to the right corresponds to all particles inducing each other. From ref. 27. Copyright Royal Academy of Sciences. | |
The first hyperpolarizability is enhanced in systems with delocalised electrons, and in particular, push–pull conjugated systems. The possibility to compute cluster hyperpolarizabilities is affected by a few factors, first of all by the computationally demanding quadratic response theory calculation, which must be performed to provide LoProp hyperpolarizabilities. Also the convergence of the iterative calculations of the induced dipole moments of all the point dipoles could be demanding. This process scales approximately as ![[scr O, script letter O]](https://www.rsc.org/images/entities/char_e52e.gif) (N2) with respect to the amount of interacting point dipoles. Finally solving the linear system of equations may take time depending on the scaling techniques applied.
(N2) with respect to the amount of interacting point dipoles. Finally solving the linear system of equations may take time depending on the scaling techniques applied.
5 Decomposition of dispersion interaction
As shown in the previous sections the LoProp scheme can be applied to different kinds of properties and intermolecular forces that describe how molecules attract or repel each other. One such force is the attractive long-range intermolecular dispersion force which in general is very subtle and weak at short ranges compared to the electrostatic, polarization or short-range terms, but which can dominate in systems which are non-polar or even lack static multipole moments. Most of the additive force fields used today69–71 include electrostatic interaction, some include polarization but most neglect the dispersion, although it is clearly essential in many circumstances, especially for weakly bound systems.72 The long-range contributions to the dispersion energy can be attributed to the vacuum fluctuation of the quantized field, as predicted by the so-called by the Casimir–Polder integral73,74 and can be viewed as “instantaneous and mutual polarization” between two particles at a distance. In practice it is almost always modelled by a Leonard-Jones potential, where the sole distant dependent parameter often is obtained by fitting to heats of vaporization or solvation energies taken from experiments. The dispersion energy is inherently included in most energy correlated ab initio electronic structure methods and various types of many-body interaction schemes, like Møller–Plesset perturbation theory and coupled cluster theory. More recently, various density functional schemes have been applied, notably the one of Stone and Misquitta that implement intermolecular contributions including the dispersion energy by the means of the SAPT(DFT) (Symmetry Adapted Perturbation Theory-Density Functional Theory). This method gives energy contributions of monomer–monomer perturbations to various degrees, including those of dispersion origin, and has been applied for a large variety of systems.75–79
In ref. 51 a new way was described to compute the two-body contribution to the dispersion energy using ab initio theory and using the philosophy of the LoProp scheme. By combining the complex polarization propagator method and the LoProp transformation, local contributions to the Casimir–Polder interaction could be obtained. The anisotropic as well as isotropic models of the dispersion energy could thereby be analyzed in dimer systems using the decomposition scheme for the dipole–dipole polarizability. We recapitulate that the linear dipole–dipole term defines the well-known C6 coefficient for the dispersion interaction:
| |  | (83) |
where

is the polarizability for site 1 calculated at an imaginary frequency i
ω. The lower indices of
α are angular momentum labels and the order
n in the
Cn coefficient is defined as
n =
t +
t′ +
u +
u′ + 2, giving the
C6 term from the linear dipole–dipole polarizability (
t =
t′ =
u =
u′ = 1). For the derivation of this equation we refer to the Theory section. The second ingredient in the scheme is the complex polarization propagator (CPP) approach by Norman and coworkers
80,81 which implements analytical response theory to calculate the isotropic
C6 dispersion coefficient. In this approach the expression of the polarizability is transformed as a function of real frequencies only to generate the real and imaginary parts
via the Cauchy moments,
80,82,83 which is possible because the general complex polarizability is a function of complex frequencies. Harzcuk
et al. thus applied the CPP approach to calculate the Casimir–Polder integral of the isotropic
C6 coefficient where the response vectors from the CPP code were used in order to obtain the distributed real polarizabilities of atoms at imaginary frequencies. That made it possible to decompose the molecular
C6 coefficients into pairs of atomic sites, giving rise to a total dispersion in a dimer system at various relative orientations. The so obtained LoProp decomposed polarizabilities are additive and sum up to the molecular polarizability, and the sum of all LoProp
Cij6 elements is thus equal to the molecular
C6 coefficient. The LoProp dispersion calculated that way could be compared to the dispersion calculated between the centers of mass of the dimers, and with high level of theory, like the above mentioned SAPT(DFT) method.
Fig. 8 illustrates basis set convergence of LoProp decomposed atomic and total molecular C6 coefficients.
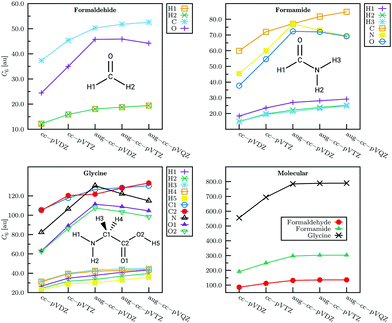 |
| | Fig. 8 Basis set convergence of LoProp decomposed atomic and total molecular C6 coefficients. From ref. 51. Copyright Royal Academy of Sciences. | |
 |
| | Fig. 9 Schematic representation of the relative orientations for the N2–N2 dimer system. From ref. 51. Copyright Royal Academy of Sciences. | |
Results for dispersion interaction energies calculated with the anisotropic models were presented and analyzed in ref. 51 for different orientations of H2, N2, CO, methane, pyridine and benzene dimers. For the diatomic dimers the LoProp results agree well with the reference values—for some (unplausible) orientations the interaction is notably underestimated. By stacking the dimers the energy is higher than for the SAPT(DFT) method but agree better at larger distances (Fig. 10 and 11). The results indicate that the leading energy term calculated from the dipole–dipole polarizability tends to underestimate the reference data for the molecular dimers, and display scatter for the methane and pyridine dimers. The convergence of the isotropic atomic and summed molecular C6 coefficients as a function of the basis is in general found satisfactory, see Fig. 8. It can be seen in the figure that the augmentation is also necessary for dispersion coefficients and consequently, polarizabilities.
 |
| | Fig. 10 Comparison of SAPT2+ results with the anisotropic dispersion energy models computed for the nitrogen dimer. Fig. 9 defines the structures. From ref. 51. Copyright Royal Academy of Sciences. | |
 |
| | Fig. 11 Dispersion energies for benzene dimers M1, S3, and S8, from left to right, respectively. SAPT(DFT) reference points were taken from ref. 84. Other results from ref. 51. Copyright Royal Academy of Sciences. | |
Some of the remaining discrepancies can probably be assigned to higher order multipole interactions, providing C8 and C10 energy terms, the inclusion of which into the decomposition scheme is a rather straightforward generalization that would improve the performance. Also the selected interaction points can be generalized to bond points. Corresponding decomposition of charges and polarizabilities have shown in some cases the importance of such a fine granulation of the interaction. Since the contribution of the dispersion energy contribution to the two-body potential is significant for large systems, and since the LoProp transformation gives transferable properties between similar groups between different systems, these results could be useful for fitting of new accurate force fields with applications to, e.g., biological DNA/protein systems. In all, the LoProp decomposition procedure as applied to dispersion interaction in ref. 51 is a promising way to design new force fields derived from ab initio reference data and to introduce localized dispersion interaction into quantum-classical hybrid methods like QM/MM methods. It may be argued that the incorporation of atomic C6 coefficients in new atomic force fields will have important ramifications in molecular dynamics studies of biomolecular systems.
6 Hyperpolarisability of collagen
The generalization of the decomposition technique to include non-linear properties has a strong motivation in the fact that non-linear optical properties of materials have progressively become more accessible and attended during recent years. That includes also biostructures and biomolecules like peptides and proteins. For such systems the inherent requirement of electron correlation in quantum mechanical methods still sets limits on the size of the systems that can be investigated at high levels of accuracy, thus promoting the need for coarser but more large-scale applicable methods to be developed.85,86 An example is the above mentioned point dipole model, originally proposed by Silberstein,35,36 and later used by Applequist et al.,37 which has found applications in prediction of molecular polarisabilities, inter-molecular potentials,87 second order susceptibilities χ(2),88 and third order susceptibilities χ(3),89 which are examples of relevant development work. It has furthermore recently been shown that the distribution of molecular properties based on the so-called “quantum theory of atoms in molecules” (QTAIM) method90 could be used for the calculation of χ(2) in crystal packings.91 Recently, Harczuk et al. showed that their derivation of decomposed non-linear polarisabilities with the LoProp-Applequist method derived in ref. 27 provides a vehicle to predict the non-linear optical characteristics of large molecular peptides and proteins.29 The capability of the method was demonstrated for the collagen triple helix, the most abundant structural protein in the human body, which in recent years has drawn attention due to its high first order non-linear response and for which second harmonic generation (SHG) imaging have been used for heart disease diagnostics and other biosensor applications.92–96 A procedure with molecular fractionation using conjugate caps in order to determine the atomic and bond contributions to the net β tensor of the collagen [(PPG)10]3 triple-helix was then employed from which the intensity of the βHRS signal and the depolarization ratios could be derived. By furthermore using Thole's exponential damping modification53,54,97 to the dyadic tensor in the Applequist equations, the hyperpolarisability could be predicted and compared with experiment. The LoProp transformation was thus used for the atom and bond decomposition of the molecular properties of each residue, in combination with the so-called MFCC98,99 procedure for cutting at the peptide bonds, which account for the properties of the helix without any long-range interactions between the residues. The second order quadratic point-dipole model is then used to calculate the inter, and intra-chain hyperpolarisation, between and inside the chains. The total scattering intensity and depolarization ratio, which are rotationally invariant and measurable, were then computed from the resulting βCollagen tensor and compared to previous experiments and calculations. This provided, for the first time, the non-linear extension of Applequist's equations to covalently bonded fragments.
Measurements of the hyper-Rayleigh scattering in collagen100 together with additive approximate models,101,102 and more advanced models as the so-called ONIOM model,103 have related the large value of β to the stacking of the peptide bonds between the (hydroxy)-proline and glycine residues. The specific rigidness caused by the [PPG] subunit has been used to explain the build up of the tensor component in the longitudinal direction of the chains, something which could not be found when modelling the [GGG] trimer. The results from an additive scheme was included, in which all the components of the dipole coupling tensor are set to zero, and the Applequist properties are the sums of the localized atomic or bond polarisabilities, and hyperpolarisabilities, respectively.
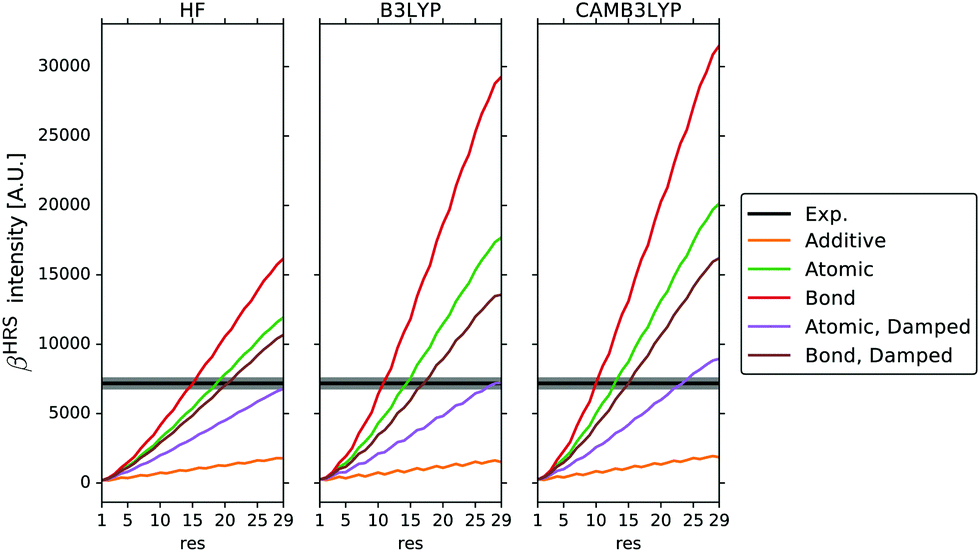
In Fig. 12, the βHRS∞ scattering intensity is plotted as a function of the amount of residues of each chain. The model peptide [(PPG)10]3 is approximately obtained with 29 residues in each chain. This collagen model has 87 residues in total, since the X-ray structure has one glycine missing in the C terminal of each chain.
 |
| | Fig. 12 The total static scattering intensity βHRS for a growing collagen triple helix. For 29 residues, the rat-tail collagen is obtained. From ref. 29. Copyright ACS Publishing. | |
The first hyperpolarisability for the amino acid constituents of the collagen triple helix was calculated employing time-dependent Hartree–Fock and density functional theory. The atomic decomposition stemming from TD-DFT/B3LYP using Thole's damping shows the best agreement with experiment. The corresponding value is for TDHF, and TD-DFT/CAMB3LYP lower, respectively, higher, see Fig. 12. The results thus show that when damping is introduced the atom + bond polarisability scheme not necessarily is better than the pure atom polarisability scheme for which the damping is parametrised. Further reasons for this are given in ref. 29. Another trend seen in Fig. 12 is that the scattering intensity grows in a slight sigmoidal shape for all the models, whereas the additive scattering, i.e. just taking the sum of the localized hyperpolarisabilities, grows linearly. Adding more residues would thus not necessarily produce a growth in intensity, but might converge with respect to the chain length. It is clear that when damping is included, the intensity is not as sensitive to the computational method used to obtain the properties.
Fig. 13 shows all the methods tested and all the models used for the depolarization ratio. It can be seen that the depolarization oscillates more wildly than the scattering intensity. The experimental value of 8.4 hints to the fact that the collagen helix is highly dipolar, with no significant octupole character. The reason why the quadratic Applequist model underestimates the dipolar nature of this collagen model might be due to that some components such as βxxx and βyyy in the collagen tensor are over-induced. It is notable that TD-B3LYP underperforms with respect to TDHF and TD-CAMB3LYP which can be attributed to the long-range exchange being important for the calculation of the non-diagonal hyperpolarizability components using the quadratic Applequist equations. Calculations in ref. 101 and 102 elucidated that the successive stacking of the π-character peptide bonds was the main reason behind the large second-order response in collagen, something that also could be confirmed in the work of Harczuk et al.29
 |
| | Fig. 13 The depolarization ratio ρ for the growing collagen triple helix. For 29 residues, the rat-tail collagen is obtained. From ref. 29. Copyright ACS Publishing. | |
A motivation for the collagen work29 can be found in the general ambition to derive models to efficiently predict properties of large biostructures with an accuracy that allows to optimize them for a particular property by structural modifications like substitutions or residual mutation.
7 Rayleigh scattering of atmospheric nanoparticles
In paper28 yet another application of the atomic decomposition scheme for polarisabilities was presented, namely for Rayleigh scattering of naturally polarised light by systems with atmospheric relevance, in particular by growing water clusters containing foreign compounds. The Rayleigh scattering intensity relates directly to the dynamical polarisability tensors of the clusters and could so be obtained by the combined LoProp-Applequist algorithm reviewed in the Theory section and in the original work.26 The study found motivation in that various particles act as cloud condensation nuclei in the atmosphere with important climate implications, like green house effect and earth's albedo,104,105 where elastic scattering of light in the atmosphere leads to cooling.
The Rayleigh scattering intensity can be obtained106 for naturally polarised light as
| | ![[scr R, script letter R]](https://www.rsc.org/images/entities/char_e531.gif) n = 45(2) + 13(Δα2) n = 45(2) + 13(Δα2) | (84) |
where the isotropic (
) and anisotropic (Δ
α) polarisabilities are defined as
| |  | (85) |
| |  | (86) |
For a spherical aerosol particle interacting with light of wavelengths approximately equal to the radius of the particle, Mie scattering constitutes the predominant scattering process, while when light interacts with aerosol particles with characteristic sizes of less than one tenth of the wavelength of the light, Rayleigh scattering guides the predominant elastic scattering effect. In Rayleigh theory, the scattering is proportional to the frequency of the incident light to the fourth power, manifesting the blue colour saturation of the sky. Thus the frequency dependent polarisability of atmospheric clusters relates to the Rayleigh scattering tensor, something that is addressable directly with the LoProp-Applequist models. It could form, as investigated in ref. 26, an excellent approach for Rayleigh scattering of particles in the nanoscale covering the scattering intensities of various incident light frequencies. Going to the other limit, to very small clusters, quantum chemistry methods can be used both for the scattering problem and for the nucleation of the clusters, a recent example being the work of Elm et al.106,107 who used quantum mechanical response theory and a combinatorial sampling approach to study the Rayleigh and hyper-Rayleigh scattering properties of some binary and ternary water clusters containing sulfuric acid and ammonia. The molecular Rayleigh, and hyper-Rayleigh, scattering intensities were expressed from the total dipole polarisability α and hyperpolarisability β tensors allowing for the exploration of the effect of cluster morphology on the scattering properties. It was found that the Rayleigh scattering intensity depends quadratically on the number of water molecules in the cluster and that a single foreign molecule (ammonia) could generate high anisotropy, which further increased the scattering intensity. The hyper Rayleigh scattering activities were found to be extremely low. That work thus gave a molecular level understanding of scattering properties of small clusters, and on the impact of foreign constituents on water clusters on Rayleigh scattering properties. As the atmosphere contains a large body of neutral molecular clusters with sizes in the nanometre scale108–111 for which the Rayleigh scattering is the dominating mechanism, it is desirable to reach the nanoscale for Rayleigh scattering simulations as achievable by the LoProp-Applequist approach.26,37 The choice in ref. 26 of atmospheric water droplets formed by adsorbed cis-pinonic acid adsorption (CPA) was motivated by the atmospheric abundance of this compound and that it is known to decrease the water droplet surface tension, leading to smaller droplet formations, causing a larger density of small droplet sizes in the clouds, which increase the overall albedo and gives a net cooling of the Earth.112 Using the LoProp-Applequist model it was then possible to scrutinise details with respect to contributions of molecular interaction, cluster size, mass constituent and dispersion on the total Rayleigh scattering. Studies were made for cluster diameters up to 5 nm, and for frequencies below the first resonance frequency, thus within the eligibility of Rayleigh scattering theory.
In Fig. 14, the increase in scattering with respect to cluster size given by the Applequist model is compared to the polarisability obtained as a summation of the polarisability components of each molecule, not taking interaction into account. All the wavelengths follow the same growth pattern, with 400 nm giving the largest increases. For the absolute magnitude of the scattering intensity the total scattering intensity for the different wavelengths depends on the cluster growth of water molecules—here the scattering intensity for all the wavelengths grow quadratically. The difference between the wavelengths also increases quadratically with respect to the static frequency. This shows that the dynamic polarisability contribution to the total scattering grows quadratically with respect to the frequency of the external field, but that the major contribution is due to the static polarisability. As shown in Fig. 15, the scattering exhibits a weak quadratic increase with respect to the concentration of CPA in the water droplet. The weak increase could be attributed to CPA avoiding the water, something which also causes it being pushed out of the water droplet sphere, residing at the surface. Close-range interactions will cause large induced polarisation, which could explain why the change in scattering intensity is almost linear with the addition of CPA to the water droplet.
 |
| | Fig. 14 The relative increase in scattering intensity from the Applequist equations for a growing droplet with different amounts of water molecules in the cluster. From ref. 28. Copyright ACS publications. | |
 |
| | Fig. 15 Absolute Rayleigh-scattering intensity for different wavelengths as a function of cis-pinonic acid mass content. From ref. 28. Copyright ACS publications. | |
In the Fig. 16, the isotropic and anisotropic contributions to the net scattering are plotted for the water only cluster, and the water cluster interacting with CPA molecules, respectively, at static frequency. It can be seen that the major contribution to the Rayleigh scattering of the pure water clusters is due to the isotropic component, which is greatly enhanced in the range of 1–1000 water molecular clusters. It is furthermore gradually increased using the Applequist equations, as compared to the non-interacting model. The anisotropic component is rather stable for the water cluster growth, but increases sharply for clusters of sizes larger than 700 water molecules. For the clusters containing 1000 water molecules and adsorbed CPA molecules, also the anisotropic component grows steadily. The Rayleigh scattering of naturally polarised light was found to increase smoothly with the increase of the overall cluster size, with the increase of mass constituent of cis-pinonic acid molecules, and with dispersion (increase of wavelength). The calculations predict that weakly adsorbed molecules may cause a lowering of the net scattering intensities compared to strongly adsorbed ones, overall affecting the albedo of clouds consisting of these types of cloud condensation nuclei.
 |
| | Fig. 16 The static isotropic and anisotropic component of the polarisability at static external field. From ref. 28. Copyright ACS publications. | |
The Applequist interaction was found to yield scattering intensities 20% larger for a cluster consisting of 1000 water molecules compared to the method where all the polarisabilities of molecules are added without interactions. It was confirmed that the scattering intensity depends quadratically on the number of water molecules in the cluster, and that it also increases quadratically with increase for the mass constituent of the foreign substance. The adsorption of the cis-pinonic acid was found to increase the contribution to the scattering intensity stemming from the anisotropic polarisability, as compared to the isotropic contribution. Paper26 indicates that large clusters with organic molecules in water is a realistic proposition for hundreds of molecules (organic and water) in the clusters and the method is shown to be both sufficiently accurate and general for any type of cluster of molecules up to size of 10 nm, which transcends already the Rayleigh scattering region and goes into Mie scattering region. That was probably the first time large clusters can be reached by a molecular-level based calculation of the Rayleigh scattering, well beyond the small ones reachable by quantum chemistry.
8 Conclusion
In the present work we have reviewed recent development of local decomposition schemes for localizing molecular properties, and highlighted their use either in QM/MM (Quantum Mechanics Molecular Mechanics) calculations or for the purpose of generating “classical” properties of large clusters. A few sample applications of such decomposition have been recapitulated and briefly discussed. The highlighted LoProp procedure was originally developed to obtain granulated solute–solvent interactions using a scheme for decomposing the quantum chemical overlap matrix and to use the resulting transformation matrix to localize a given property into a predefined level of localization. Recent extensions, highlighted in this review, of the technique comprise locally decomposed hyperpolarisabilities,27 frequency dependent polarisabilities,26 and locally decomposed frequency dependent complex polarisabilities.51 The LoProp scheme has become instrumental for many solvent interaction models, and in particular for implementations of QM/MM (quantum mechanics molecular mechanics) methods. In addition, the LoProp technique has recently been shown to be a powerful tool for building up properties of large molecular clusters, like for Rayleigh scattering of atmospheric nanoparticles28 and hyperpolarisabilities (second harmonic generation) of proteins.29 The flexibility of the model to address localized interactions with atomically decomposed charges and polarisabilities has thus been critical for the accuracy of many structure and property calculations by means of QM/MM. The flexibility not only concerns the position of the interaction points but also the multipolar expansion of charges and polarisabilities. By means of LoProp several systematically connected models for the solvent interaction can be formulated, ranging from simple molecular point charges to bond and atom centred multipole expanded charges and polarisabilities. The precision needed, and thus the level of decomposition, is to a large extent dictated by the property that is to be calculated. The level of decomposition also has a bearing on the standard problem how to split up the QM and MM parts of the system in a QM/MM calculation, for instance, the need to bring in water molecules in the QM part is often relieved by a careful LoProp MM parametrisation of water.3 To the known limitations of the LoProp method one can mention the tendency to “overpolarise” the interaction, especially to the QM part close to the QM-MM interface but also between closely encountering MM molecules. The (ad hoc) introduction of distributed point charges and polarisabilities by Gaussian broadenings remedies the problem to some extent.16,53 It goes without saying that the LoProp model does not handle charge transfer unless special parametrization for that is derived (e.g. in terms of capacitance parameters as in the QMCMM model mentioned above).
The flexibility of the LoProp method makes it open ended for further fine-tuning of accuracy, thus to multipole expand charges and polarisabilities, and include higher order dispersion coefficients, which is straightforwardly doable if the underlying property integrals are at hand. Such extensions would be the most natural measure of development of the present implementations in order to reach the ultimate precision of such a decomposition scheme. These implementations are in principle possible to introduce into QM/MM, although, to our knowledge, such a QM/MM has not yet been theoretically formulated for localized dispersion. Importantly, the presented scheme would allow to develop chemically dependent local dispersion interaction force fields in molecular dynamics, improving the commonly used force fields of atomically dependent Lennard-Jones interactions. It is desirable to “homogenize” the force fields utilized in the so-called integrated approach that combines MD with QM/MM, thus that the force fields in the two types of calculations are identical. Implementation of the schemes reviewed here into general MD simulation software would certainly not be straightforward “add-on” efforts, as current MD programs often are heavily parametrised towards certain reference data. The field of decomposition of molecular properties is thus wide open for further research, and important new applications for cluster properties, QM/MM and molecular dynamics can be envisaged.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors would like to acknowledge financial support from the Swedish Research Council: 2014-05270 (OV), and 2016-03619 (HÅ). The computational resources were provided by Swedish National Infrastructure for Computing (SNIC) at the National Supercomputing Centre (NSC) and the High Performance Computing Centre North (HPC2N), projects “Multiphysics Modeling of Molecular Materials”, SNIC 023/07-18, and “Design of Force Fields for Theoretical Spectroscopy”, SNIC-2015-1-230.
References
- C. B. Nielsen, O. Christiansen, K. V. Mikkelsen and J. Kongsted, J. Chem. Phys., 2007, 126, 154112 CrossRef PubMed.
- Z. Rinkevicius, N. A. Murugan, J. Kongsted, B. Frecus, A. H. Steindal and H. Agren, J. Chem. Theory Comput., 2011, 7, 3261–3271 CrossRef CAS PubMed.
- Z. Rinkevicius, N. A. Murugan, J. Kongsted, K. Aidas, A. H. Steindal and H. Agren, J. Phys. Chem. B, 2011, 115, 4350–4358 CrossRef CAS PubMed.
- A. H. Steindal, K. Ruud, L. Frediani, K. Aidas and J. Kongsted, J. Phys. Chem. B, 2011, 115, 3027–3037 CrossRef CAS PubMed.
- M. N. Pedersen, E. D. Hedegård, J. M. H. Olsen, J. Kauczor, P. Norman and J. Kongsted, J. Chem. Theory Comput., 2014, 10, 1164–1171 CrossRef CAS PubMed.
- H. M. Senn and W. Thiel, Angew. Chem., Int. Ed., 2009, 48, 1198–1229 CrossRef CAS PubMed.
- L. Adamowic, M. A. Freitag and M. S. Gordon, J. Chem. Phys., 2003, 118, 6725–6732 CrossRef.
- I. V. R. Claudia, F. Jonathan and L. V. Slipchenko, J. Chem. Phys., 2018, 149, 094103 CrossRef PubMed.
- L. V. Slipchenko, M. S. Gordon and K. Ruedenberg, J. Phys. Chem. A, 2017, 121, 9495–9507 CrossRef CAS PubMed.
- Z. Rinkevicius, X. Li, J. A. R. Sandberg and H. Ågren, Phys. Chem. Chem. Phys., 2014, 16, 661–665 RSC.
- A. J. Stone, J. Chem. Theory Comput., 2005, 1, 1128–1132 CrossRef CAS PubMed.
- E. R. Kjellgren, J. M. Haugaard Olsen and J. Kongsted, J. Chem. Theory Comput., 2018, 14, 4309–4319 CrossRef CAS PubMed.
- M. Scheurer, M. F. Herbst, P. Reinholdt, J. M. Haugaard Olsen, A. Dreuw and J. Kongsted, J. Chem. Theory Comput., 2018, 14, 4870–4883 CrossRef CAS PubMed.
- M. Scheurer, M. F. Herbst, P. Reinholdt, J. M. H. Olsen, A. Dreuw and J. Kongsted, J. Chem. Theory Comput., 2018, 14, 4870–4883 CrossRef CAS PubMed.
- Z. Rinkevicius, B. Frecus, O. Vahtras, J. Kongsted and H. Ågren, J. Phys. Chem. B, 2012, 8, 257–263 CAS.
- Z. Rinkevicius, X. Li, J. A. R. Sandberg, K. V. Mikkelsen and H. Ågren, J. Chem. Theory Comput., 2014, 10, 989–1003 CrossRef CAS PubMed.
- F. Iori and S. Corni, J. Comput. Chem., 2008, 29, 1656–1666 CrossRef CAS PubMed.
- F. Iori, R. Di Felice, E. Molinari and S. Corni, J. Comput. Chem., 2009, 30, 1465–1476 CrossRef CAS PubMed.
- O. Crescenzi, M. Pavone, F. De Angelis and V. Barone, J. Phys. Chem. B, 2004, 109, 445–453 CrossRef PubMed.
- L. Gagliardi, R. Lindh and G. Karlström, J. Chem. Phys., 2004, 121, 4494–4500 CrossRef CAS PubMed.
- N. A. Murugan, J. Kongsted, Z. Rinkevicius and H. Ågren, Phys. Chem. Chem. Phys., 2012, 14, 1107 RSC.
- K. Aidas, A. Møgelhøj, C. B. Nielsen, K. V. Mikkelsen, K. Ruud, O. Christiansen and J. Kongsted, J. Phys. Chem. A, 2007, 111, 4199–4210 CrossRef CAS PubMed.
- J. J. Eriksen, J. M.
H. Olsen, K. Aidas, H. Ågren, K. V. Mikkelsen and J. Kongsted, J. Comput. Chem., 2011, 32, 2853–2864 CrossRef CAS PubMed.
- T. Löytynoja, I. Harczuk, K. Jänkälä, O. Vahtras and H. Agren, J. Chem. Phys., 2017, 146, 124902 CrossRef PubMed.
- I. Harczuk, N. A. Murugan, O. Vahtras and H. Ågren, J. Chem. Theory Comput., 2014, 10, 3492–3502 CrossRef CAS PubMed.
- I. Harczuk, O. Vahtras and H. Ågren, Phys. Chem. Chem. Phys., 2015, 17, 7800–7812 RSC.
- I. Harczuk, O. Vahtras and H. Ågren, Phys. Chem. Chem. Phys., 2016, 18, 8710–8722 RSC.
- I. Harczuk, O. Vahtras and H. Ågren, J. Phys. Chem. B, 2016, 120, 4296–4301 CrossRef CAS PubMed.
- I. Harczuk, O. Vahtras and H. Ågren, J. Phys. Chem. Lett., 2016, 7, 2132–2138 CrossRef CAS PubMed.
- J. Olsen and P. Jørgensen, J. Chem. Phys., 1985, 82, 3235–3264 CrossRef CAS.
- P. Sałek, O. Vahtras, T. Helgaker and H. Ågren, J. Chem. Phys., 2002, 117, 9630–9645 CrossRef.
- K. Aidas, C. Angeli, K. L. Bak, V. Bakken, R. Bast, L. B. O. Christiansen, R. Cimiraglia, S. Coriani, P. Dahle, E. K. Dalskov, U. E. T. Enevoldsen, J. J. Eriksen, P. Ettenhuber, B. Fernández, L. Ferrighi, H. F. L. Frediani, K. Hald, A. Halkier, C. Hättig, H. Heiberg, T. Helgaker, A. C. Hennum, H. Hettema, E. Hjertenæs, S. Høst, I. M. Høyvik, M. F. Iozzi, B. Jansík, H. J. A. Jensen, D. Jonsson, P. Jørgensen, J. Kauczor, S. Kirpekar, T. Kjærgaard, W. Klopper, S. Knecht, R. K. H. Koch, J. Kongsted, A. Krapp, K. Kristensen, A. Ligabue, O. B. Lutnæs, J. I. Melo, K. V. Mikkelsen, R. H. Myhre, C. Neiss, C. B. Nielsen, P. Norman, J. Olsen, M. H. Olsen, A. Osted, M. J. Packer, F. Pawlowski, T. B. Pedersen, P. F. Provasi, S. Reine, Z. Rinkevicius, T. A. Ruden, R. K. Ruud, V. V. Rybkin, P. Sałek, C. C. Samson, A. S. de Merás, T. Saue, S. P. A. Sauer, B. Schimmelpfennig, K. Sneskov, A. H. Stendal, K. O. Sylvester-Hvid, P. R. Taylor, A. M. Teale, E. I. Tellgren, D. P. Tew, A. J. Thorvaldsen, L. Thøgersen, O. Vahtras, M. A. Watson, D. J. D. Wilson, M. Ziolkowski and H. Ågren, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2014, 4, 269–284 CAS.
- P. Sałek, O. Vahtras, T. Helgaker and H. Ågren, J. Chem. Phys., 2002, 117, 9630 CrossRef.
- J. Applequist, J. Chem. Phys., 1985, 83, 809–826 CrossRef CAS.
- L. Silberstein, Philos. Mag., 1917, 33, 92–128 CAS.
- L. Silberstein, Philos. Mag., 1917, 33, 521–533 CAS.
- J. Applequist, J. R. Carl and K.-K. Fung, J. Am. Chem. Soc., 1972, 94, 2952–2960 CrossRef CAS.
- J. Applequist, J. Chem. Phys., 1993, 98, 7664 CrossRef CAS.
- L. Jensen, P. O. Åstrand, A. Osted, J. Kongsted and K. V. Mikkelsen, J. Chem. Phys., 2002, 116, 4001–4010 CrossRef CAS.
- T. Hansen, L. Jensen, P. O. Åstrand and K. V. Mikkelsen, J. Chem. Theory Comput., 2005, 1, 626–633 CrossRef CAS PubMed.
- L. Jensen, K. O. Sylvester-Hvid, K. V. Mikkelsen and P.-O. Åstrand, J. Phys. Chem. A, 2003, 107, 2270–2276 CrossRef CAS.
- L. Jensen, P.-O. Åstrand and K. V. Mikkelsen, Nano Lett., 2003, 3, 661–665 CrossRef CAS.
- C. E. Dykstra, J. Comput. Chem., 1988, 9, 476–487 CrossRef CAS.
- J. D. Augspurger and C. E. Dykstra, J. Comput. Chem., 1992, 43, 135–146 CAS.
- J. M. Stout and C. E. Dykstra, J. Am. Chem. Soc., 1995, 117, 5127–5132 CrossRef CAS.
- C. E. Dykstra, Chem. Phys. Lett., 1999, 305, 132 CrossRef.
- R. W. Munn, Mol. Phys., 1988, 64, 1 CrossRef.
- M. in het Panhuis and R. W. Munn, J. Chem. Phys., 2000, 113, 10685 CrossRef CAS.
-
O. Vahtras, LoProp for Dalton, 2014, DOI:10.5281/zenodo.13276.
- V. Magnasco, G. Figari and C. Costa, THEOCHEM, 1992, 261, 237–253 CrossRef.
- I. Harczuk, B. Nagy, F. Jensen, O. Vahtras and H. Ågren, Phys. Chem. Chem. Phys., 2017, 19, 20241–20250 RSC.
-
A. Salam, Appendix B: Rotational Averaging of Cartesian Tensors, John Wiley & Sons, Inc., 2009, pp. 378–380 Search PubMed.
- B. Thole, Chem. Phys., 1981, 59, 341–350 CrossRef CAS.
- P. T. Van Duijnen and M. Swart, J. Phys. Chem. A, 1998, 102, 2399–2407 CrossRef CAS.
- H. E. Watson and K. L. Ramaswamy, Proc. R. Soc. London, Ser. A, 1936, 156, 144–157 CAS.
- P. Ren and J. W. Ponder, J. Comput. Chem., 2002, 23, 1497–1506 CrossRef CAS PubMed.
- P. Ren and J. W. Ponder, J. Phys. Chem. B, 2003, 107, 5933–5947 CrossRef CAS.
- J. Wang, P. Cieplak, J. Li, T. Hou, R. Luo and Y. Duan, J. Phys. Chem. B, 2011, 115, 3091–3099 CrossRef CAS PubMed.
- J. Wang, P. Cieplak, J. Li, J. Wang, Q. Cai, M. Hsieh, H. Lei, R. Luo and Y. Duan, J. Phys. Chem. B, 2011, 115, 3100–3111 CrossRef CAS PubMed.
- R. Bosque and J. Sales, J. Chem. Inf. Comput. Sci., 2002, 42, 1154–1163 CrossRef CAS PubMed.
- K. Aidas, A. Møgelhøj, E. J. K. Nilsson, M. S. Johnson, K. V. Mikkelsen, O. Christiansen, P. Söderhjelm and J. Kongsted, J. Chem. Phys., 2008, 128, 194503 CrossRef PubMed.
- Z. Rinkevicius, N. A. Murugan, J. Kongsted, K. Aidas, A. H. Steindal and H. Ågren, J. Phys. Chem. B, 2011, 115, 4350–4358 CrossRef CAS PubMed.
- M. S. Norby, O. Vahtras, P. Norman and J. Kongsted, Mol. Phys., 2017, 115, 39–47 CrossRef.
- J. F. Ward and C. K. Miller, Phys. Rev. A: At., Mol., Opt. Phys., 1979, 19, 826 CrossRef CAS.
- B. F. Levine, J. Chem. Phys., 1976, 65, 2429 CrossRef CAS.
- K. V. Mikkelsen, Y. Luo, H. Ågren and P. Jørgensen, J. Chem. Phys., 1995, 102, 9362 CrossRef CAS.
- K. O. Sylvester-Hvid, K. V. Mikkelsen, P. Norman, D. Jonsson and H. Ågren, J. Phys. Chem. A, 2004, 108, 8961–8965 CrossRef CAS.
- A. H. Narten, J. Chem. Phys., 1971, 55, 2263 CrossRef CAS.
- K. Vanommeslaeghe, E. Hatcher, C. Acharya, S. Kundu, S. Zhong, J. Shim, E. Darian, O. Guvench, P. Lopes, I. Vorobyov and A. D. Mackerell, J. Comput. Chem., 2010, 31, 671–690 CAS.
-
D. Case, T. Darden, T. Cheatham, III, C. Simmerling, J. Wang, R. Duke, R. Luo, R. Walker, W. Zhang, K. Merz, B. Roberts, S. Hayik, A. Roitberg, G. Seabra, J. Swails, A. Goetz, I. Kolossváry, K. Wong, F. Paesani, J. Vanicek, R. Wolf, J. Liu, X. Wu, S. Brozell, T. Steinbrecher, H. Gohlke, Q. Cai, X. Ye, J. Wang, M.-J. Hsieh, G. Cui, D. Roe, D. Mathews, M. Seetin, R. Salomon-Ferrer, C. Sagui, V. Babin, T. Luchko, S. Gusarov, A. Kovalenko and P. Kollman, Amber 12, University of California, San Fransisco, 2012 Search PubMed.
- W. L. Jorgensen and J. Tirado-Rives, J. Am. Chem. Soc., 1988, 110, 1657–1666 CrossRef CAS PubMed.
- J.-P. Piquemal, L. Perera, G. A. Cisneros, P. Ren, L. G. Pedersen and T. A. Darden, J. Chem. Phys., 2006, 125, 054511 CrossRef.
- H. Casimir and D. Polder, Phys. Rev., 1948, 73, 360 CrossRef CAS.
- E. Power and T. Thirunamachandran, Phys. Rev. A: At., Mol., Opt. Phys., 1993, 48, 4761 CrossRef CAS.
- A. J. Stone and A. J. Misquitta, Int. Rev. Phys. Chem., 2007, 26, 193–222 Search PubMed.
- R. Z. Khaliullin, E. A. Cobar, R. C. Lochan, A. T. Bell and M. Head-Gordon, J. Phys. Chem. A, 2007, 111, 8753–8765 CrossRef CAS PubMed.
- R. J. Azar and M. Head-Gordon, J. Chem. Phys., 2012, 136, 024103 CrossRef PubMed.
- A. J. Misquitta, R. Podeszwa, B. Jeziorski and K. Szalewicz, J. Chem. Phys., 2005, 123, 214103 CrossRef PubMed.
- A. J. Misquitta and A. J. Stone, Mol. Phys., 2008, 106, 1631–1643 CrossRef CAS.
- P. Norman, D. M. Bishop, H. J. Aa. Jensen and J. Oddershede, J. Chem. Phys., 2001, 115, 10323–10334 CrossRef CAS.
- P. Norman, D. M. Bishop, H. J. Aa. Jensen and J. Oddershede, J. Chem. Phys., 2005, 123, 194103 CrossRef PubMed.
- A. Jiemchooroj, P. Norman and B. E. Sernelius, J. Chem. Phys., 2005, 123, 124312 CrossRef PubMed.
- A. Jiemchooroj, P. Norman and B. E. Sernelius, J. Chem. Phys., 2006, 125, 124306 CrossRef PubMed.
- R. Podeszwa, R. Bukowski and K. Szalewicz, J. Phys. Chem. A, 2006, 110, 10345–10354 CrossRef CAS PubMed.
- G. Maroulis, J. Chem. Phys., 1998, 108, 5432–5448 CrossRef CAS.
- B. Champagne, E. Botek, M. Nakano, T. Nitta and K. Yamaguchi, J. Chem. Phys., 2005, 122, 114315 CrossRef PubMed.
- C. E. Dykstra, J. Comput. Chem., 1988, 9, 476–487 CrossRef CAS.
- R. Munn, Int. J. Quantum Chem., 1992, 43, 159–169 CrossRef CAS.
- H. Reis, M. Papadopoulos, P. Calaminici, K. Jug and A. Koster, Chem. Phys., 2000, 261, 359–371 CrossRef CAS.
- R. F. W. Bader and T. T. Nguyendang, Adv. Quantum Chem., 1981, 14, 63–124 CrossRef CAS.
- T. Seidler and B. Champagne, Phys. Chem. Chem. Phys., 2015, 17, 19546–19556 RSC.
- S. Roth and I. Freund, J. Chem. Phys., 1979, 70, 1637–1643 CrossRef CAS.
- I. Freund, M. Deutsch and A. Sprecher, Biophys. J., 1986, 50, 693–712 CrossRef CAS PubMed.
- E. Brown, T. McKee, A. Pluen, B. Seed, Y. Boucher and R. K. Jain,
et al.
, Nat. Med., 2003, 9, 796–800 CrossRef CAS PubMed.
- G. Cox, E. Kable, A. Jones, I. Fraser, F. Manconi and M. D. Gorrell, J. Struct. Biol., 2003, 141, 53–62 CrossRef CAS PubMed.
- A. C. Ko, A. Ridsdale, M. S. Smith, L. B. Mostaco-Guidolin, M. D. Hewko, A. F. Pegoraro, E. K. Kohlenberg, B. Schattka, M. Shiomi and A. Stolow,
et al.
, J. Biomed. Opt., 2010, 15, 020501 CrossRef PubMed.
- M. Swart and P. T. van Duijnen, Mol. Simul., 2006, 32, 471–484 CrossRef CAS.
- D. W. Zhang and J. Z. H. Zhang, J. Chem. Phys., 2003, 119, 3599 CrossRef CAS.
- P. Söderhjelm and U. Ryde, J. Phys. Chem. A, 2009, 113, 617–627 CrossRef PubMed.
- A. Deniset-Besseau, J. Duboisset, E. Benichou, F. Hache, P.-F. Brevet and M.-C. Schanne-Klein, J. Phys. Chem. B, 2009, 113, 13437–13445 CrossRef CAS PubMed.
- C. Loison and D. Simon, J. Phys. Chem. A, 2010, 114, 7769–7779 CrossRef CAS PubMed.
- A. E. Tuer, S. Krouglov, N. Prent, R. Cisek, D. Sandkuijl, K. Yasufuku, B. C. Wilson and V. Barzda, J. Phys. Chem. B, 2011, 115, 12759–12769 CrossRef CAS PubMed.
- M. de Wergifosse, J. de Ruyck and B. Champagne, J. Phys. Chem. C, 2014, 118, 8595–8602 CrossRef CAS.
- A. Dai, Wiley Interdiscip. Rev.: Clim. Change, 2011, 2, 45–65 Search PubMed.
-
V. F. McNeill, N. Sareen and A. N. Schwier, Atmospheric and Aerosol Chemistry, Springer, 2014, pp. 201–259 Search PubMed.
- J. Elm, P. Norman, M. Bilde and K. V. Mikkelsen, Phys. Chem. Chem. Phys., 2014, 16, 10883 RSC.
- J. Elm, P. Norman and K. V. Mikkelsen, Phys. Chem. Chem. Phys., 2015, 17, 15701–15709 RSC.
- M. Kulmala, I. Riipinen, M. Sipila, H. E. Manninen, T. Petaja, H. Junninen, M. Dal Maso, G. Mordas, A. Mirme, M. Vana, A. Hirsikko, L. Laakso, R. M. Harrison, I. Hanson, C. Leung, K. E. J. Lehtinen and V.-M. Kerminen, Science, 2007, 318, 89–92 CrossRef CAS PubMed.
- M. Sipila, K. Lehtipalo, M. Kulmala, T. Petaja, H. Junninen, P. P. Aalto, H. E. Manninen, E. M. Kyro, E. Asmi, I. Riipinen, J. Curtius, A. Kuerten, S. Borrmann and C. D. O'Dowd, Atmos. Chem. Phys., 2008, 8, 4049–4060 CrossRef CAS.
- K. Lehtipalo, M. Sipila, I. Riipinen, T. Nieminen and M. Kulmala, Atmos. Chem. Phys., 2009, 9, 4177–4184 CrossRef CAS.
- H. E. Manninen, T. Petaja, E. Asmi, I. Riipinen, T. Nieminen, J. Mikkila, U. Horrak, A. Mirme, S. Mirme, L. Laakso, V.-M. Kerminen and M. Kulmala, Boreal Environ. Res., 2009, 14, 591–605 CAS.
- X. Li, T. Hede, Y. Tu, C. Leck and H. Ågren, J. Phys. Chem. Lett., 2010, 1, 769–773 CrossRef CAS.
|
| This journal is © the Owner Societies 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *ab,
Ignat
Harczuk
a and
Olav
Vahtras
*ab,
Ignat
Harczuk
a and
Olav
Vahtras
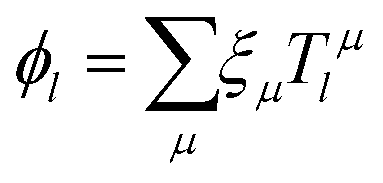



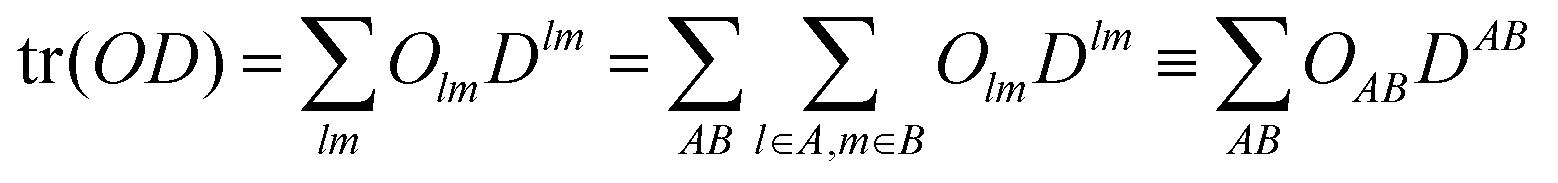


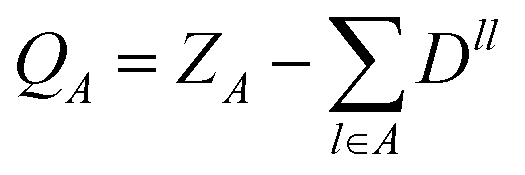
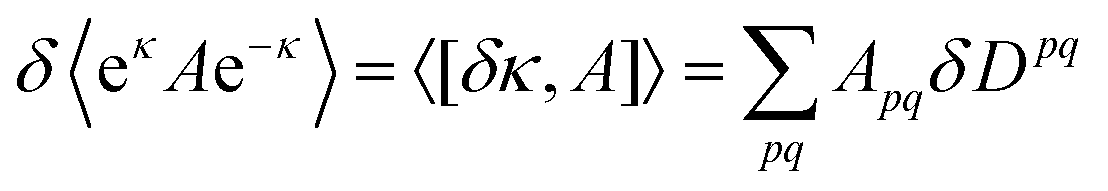



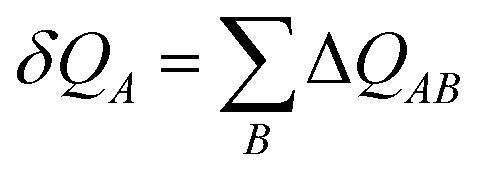
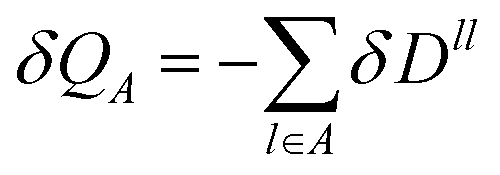





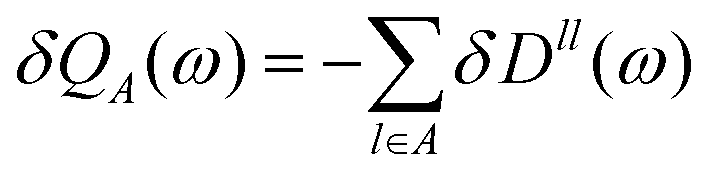
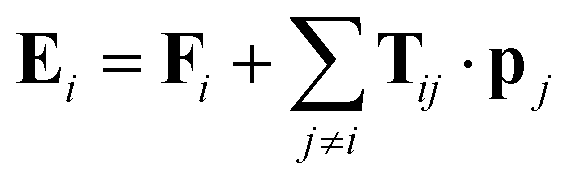
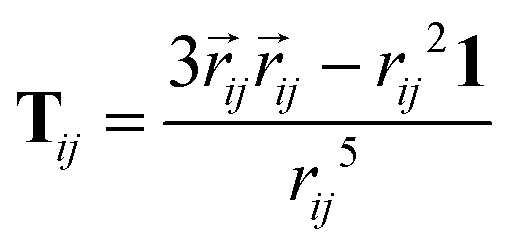

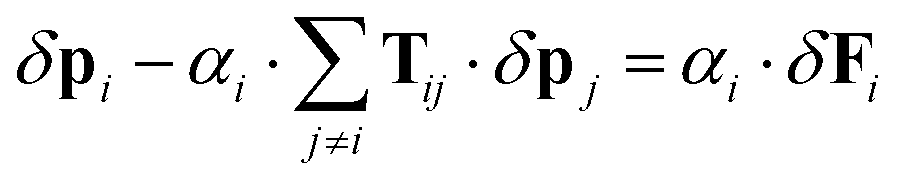







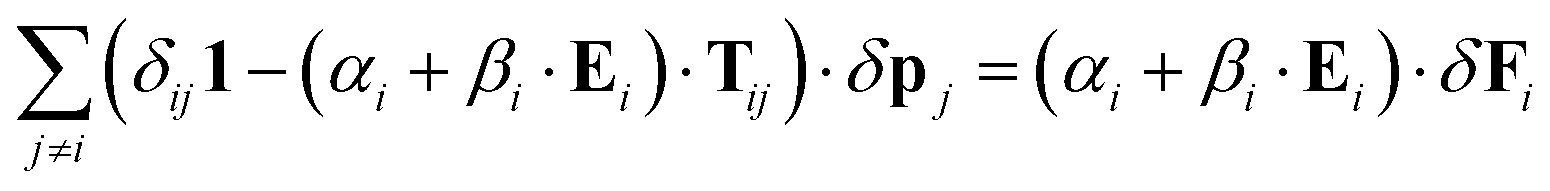










 , (the latter corresponding to the dipole interaction between the molecule and an external electric field with frequency ω) we can extract the polarizability as the linear response function
, (the latter corresponding to the dipole interaction between the molecule and an external electric field with frequency ω) we can extract the polarizability as the linear response function



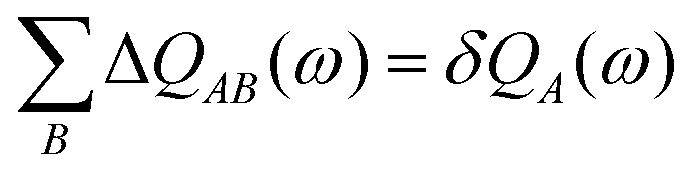








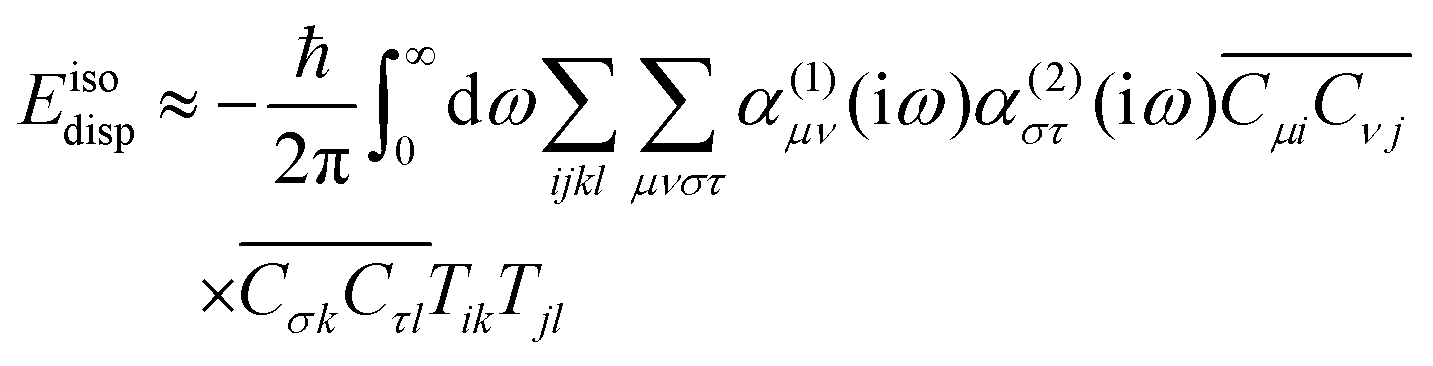


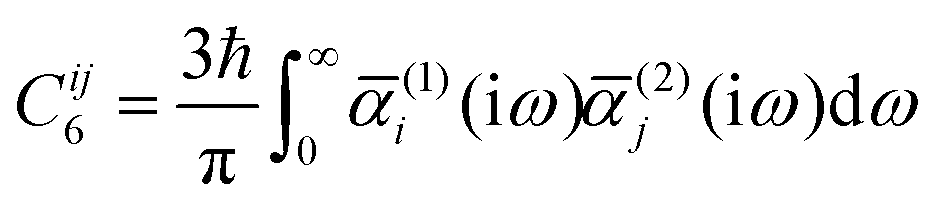
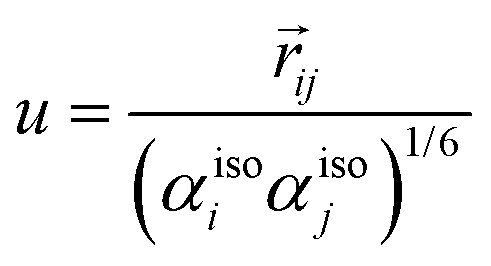




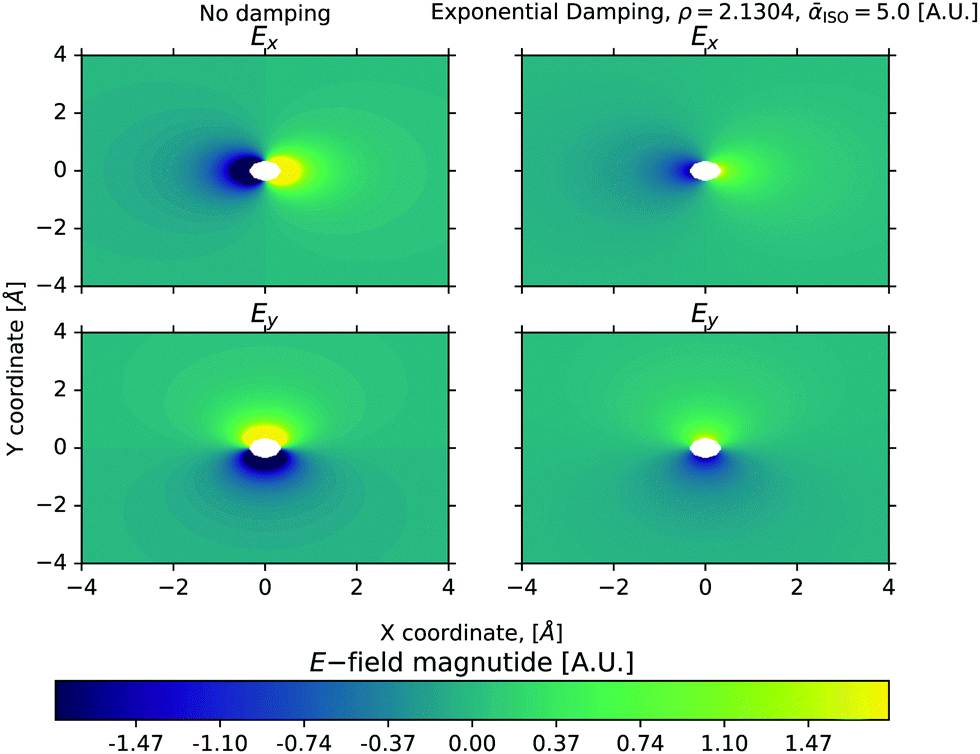

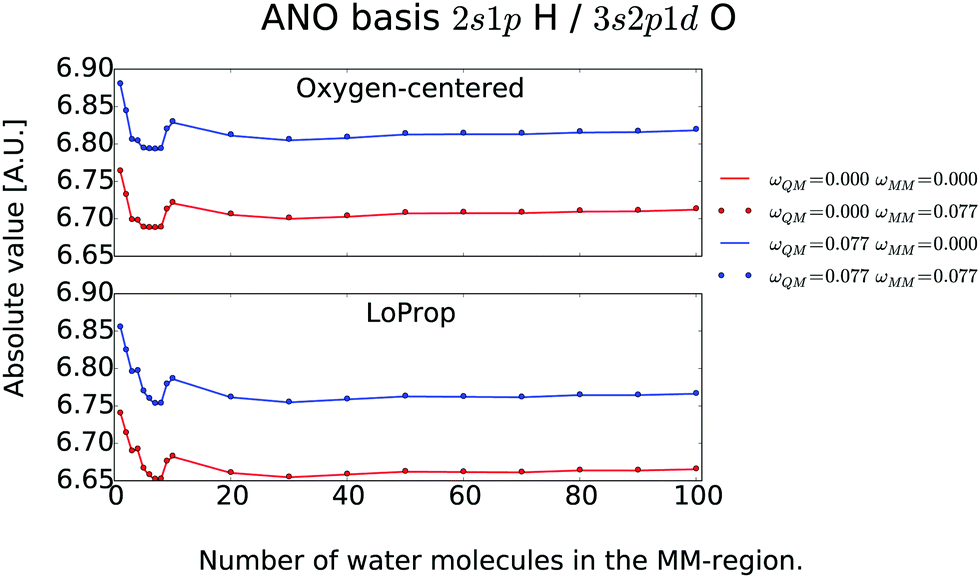





 is the polarizability for site 1 calculated at an imaginary frequency iω. The lower indices of α are angular momentum labels and the order n in the Cn coefficient is defined as n = t + t′ + u + u′ + 2, giving the C6 term from the linear dipole–dipole polarizability (t = t′ = u = u′ = 1). For the derivation of this equation we refer to the Theory section. The second ingredient in the scheme is the complex polarization propagator (CPP) approach by Norman and coworkers80,81 which implements analytical response theory to calculate the isotropic C6 dispersion coefficient. In this approach the expression of the polarizability is transformed as a function of real frequencies only to generate the real and imaginary parts via the Cauchy moments,80,82,83 which is possible because the general complex polarizability is a function of complex frequencies. Harzcuk et al. thus applied the CPP approach to calculate the Casimir–Polder integral of the isotropic C6 coefficient where the response vectors from the CPP code were used in order to obtain the distributed real polarizabilities of atoms at imaginary frequencies. That made it possible to decompose the molecular C6 coefficients into pairs of atomic sites, giving rise to a total dispersion in a dimer system at various relative orientations. The so obtained LoProp decomposed polarizabilities are additive and sum up to the molecular polarizability, and the sum of all LoProp Cij6 elements is thus equal to the molecular C6 coefficient. The LoProp dispersion calculated that way could be compared to the dispersion calculated between the centers of mass of the dimers, and with high level of theory, like the above mentioned SAPT(DFT) method.
is the polarizability for site 1 calculated at an imaginary frequency iω. The lower indices of α are angular momentum labels and the order n in the Cn coefficient is defined as n = t + t′ + u + u′ + 2, giving the C6 term from the linear dipole–dipole polarizability (t = t′ = u = u′ = 1). For the derivation of this equation we refer to the Theory section. The second ingredient in the scheme is the complex polarization propagator (CPP) approach by Norman and coworkers80,81 which implements analytical response theory to calculate the isotropic C6 dispersion coefficient. In this approach the expression of the polarizability is transformed as a function of real frequencies only to generate the real and imaginary parts via the Cauchy moments,80,82,83 which is possible because the general complex polarizability is a function of complex frequencies. Harzcuk et al. thus applied the CPP approach to calculate the Casimir–Polder integral of the isotropic C6 coefficient where the response vectors from the CPP code were used in order to obtain the distributed real polarizabilities of atoms at imaginary frequencies. That made it possible to decompose the molecular C6 coefficients into pairs of atomic sites, giving rise to a total dispersion in a dimer system at various relative orientations. The so obtained LoProp decomposed polarizabilities are additive and sum up to the molecular polarizability, and the sum of all LoProp Cij6 elements is thus equal to the molecular C6 coefficient. The LoProp dispersion calculated that way could be compared to the dispersion calculated between the centers of mass of the dimers, and with high level of theory, like the above mentioned SAPT(DFT) method.