
The HKrCCH⋯CO2 complex: an ab initio and matrix-isolation study†
 ab
Vladimir I.
Feldman
*b
and
ab
Vladimir I.
Feldman
*b
and
Abstract
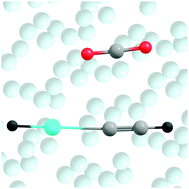
We report an experimental and theoretical study on new noble-gas hydride complex HKrCCH⋯CO2, which is the first known complex of a krypton hydride with carbon dioxide. This species was prepared by the annealing-induced H + Kr + CCH⋯CO2 reaction in a krypton matrix, the CCH⋯CO2 complexes being produced by UV photolysis of propiolic acid (HCCCOOH). The H–Kr stretching mode of the HKrCCH⋯CO2 complex at 1316 cm−1 is blue-shifted by 74 cm−1 from the most intense H–Kr stretching band of HKrCCH monomer. The observed blue shift indicates the stabilization of the H–Kr bond upon complexation, which is characteristic of complexes of noble-gas hydrides. This spectral shift is slightly larger than that of the HKrCCH⋯C2H2 complex (+60 cm−1) and significantly larger than that of the HXeCCH⋯CO2 complex (+32 and +6 cm−1). On the basis of comparison with ab initio computations at the MP2 and CCSD(T) levels of theory, the experimentally observed absorption is assigned to the quasi-parallel configuration of the HKrCCH⋯CO2 complex. The calculated complexation-induced spectral shift of the H–Kr stretching band (60.4 or 72.7 cm−1 from the harmonic calculations at the MP2 and CCSD(T) levels, respectively) agrees well with the experimental value.
- This article is part of the themed collection: Challenges in spectroscopy: accuracy vs interpretation from isolated molecules to condensed phases
 Please wait while we load your content...
Please wait while we load your content...

