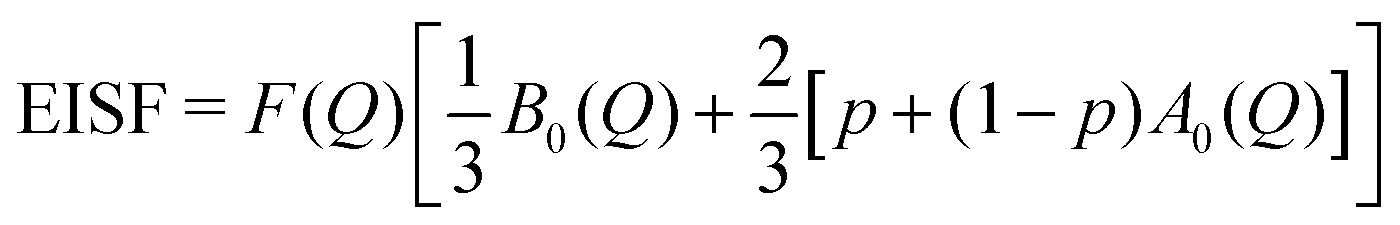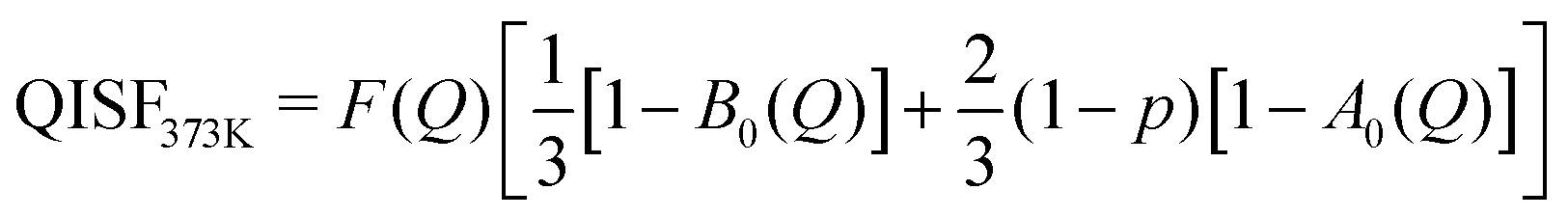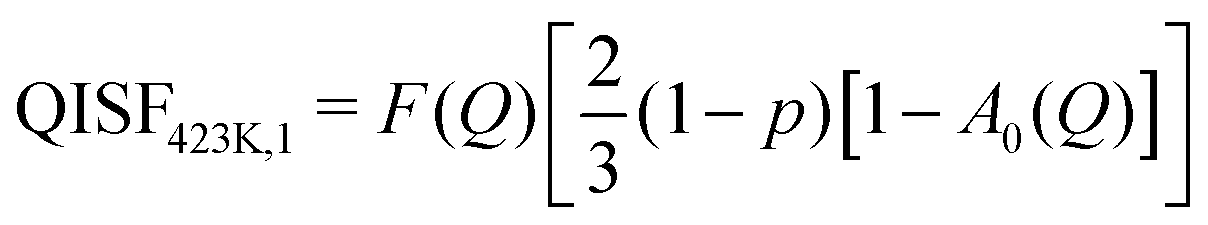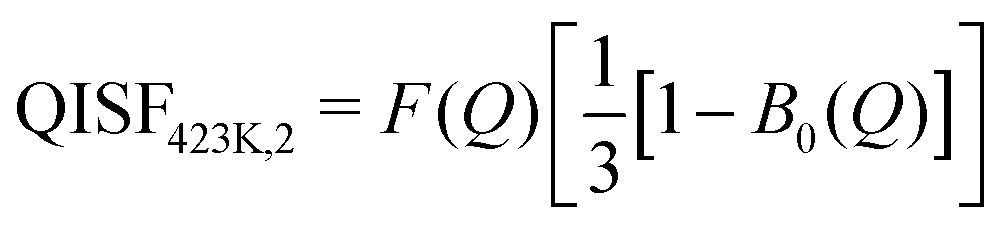A quasielastic and inelastic neutron scattering study of the alkaline and alkaline-earth borohydrides LiBH4 and Mg(BH4)2 and the mixture LiBH4 + Mg(BH4)2†
Received
9th July 2018
, Accepted 3rd December 2018
First published on 4th December 2018
Abstract
Quasielastic neutron scattering was used to investigate the low energy transfer dynamics of the complex borohydrides Mg(BH4)2 in the α- and β-modifications, LiBH4 in the low and high temperature crystal structure, and an 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar mixture of LiBH4 + α-Mg(BH4)2. All investigated compounds show a rich dynamic behaviour below an energy range of ΔE = 10 meV with the superposition of rotational dynamics of the constituent [BH4]− anions and low lying lattice modes. For Mg(BH4)2, the rotational diffusion of the [BH4] units was found to be much more activated in the metastable β-polymorph compared to the α-phase, and the low lying lattice modes are even softer in the former crystal structure. In Mg(BH4)2, the structural phase transition is mainly governed by the lattice dynamics, while alkaline LiBH4 exhibits a transition of the [BH4] rotations around the phase transition temperature. Ball milled LiBH4 + α-Mg(BH4)2 remains a physical mixture of the parent compounds and each component retains its characteristic dynamic signature up to the melting temperature.
:1 molar mixture of LiBH4 + α-Mg(BH4)2. All investigated compounds show a rich dynamic behaviour below an energy range of ΔE = 10 meV with the superposition of rotational dynamics of the constituent [BH4]− anions and low lying lattice modes. For Mg(BH4)2, the rotational diffusion of the [BH4] units was found to be much more activated in the metastable β-polymorph compared to the α-phase, and the low lying lattice modes are even softer in the former crystal structure. In Mg(BH4)2, the structural phase transition is mainly governed by the lattice dynamics, while alkaline LiBH4 exhibits a transition of the [BH4] rotations around the phase transition temperature. Ball milled LiBH4 + α-Mg(BH4)2 remains a physical mixture of the parent compounds and each component retains its characteristic dynamic signature up to the melting temperature.
1 Introduction
Complex tetraborohydrides are a class of materials that have been studied intensely for their high volumetric and gravimetric hydrogen content and their potential use in solid state hydrogen storage applications. However, high thermodynamic stability, sluggish kinetics, formation of by-products and poor reversibility make these materials still unsuitable for viable storage systems. Among the complex hydrides, the most investigated compounds are lithium-, magnesium-, and calcium borohydrides which exhibit high hydrogen contents of 18.4 wt% H2 (LiBH4), 14.9 wt% H2 (Mg(BH4)2), and 11.6 wt% H2 (Ca(BH4)2), respectively.1–5 In general, complex hydrides are salt like materials, composed of [BH4]− anions where four hydrogen atoms are covalently bound around the central boron atom, and the positively charged alkaline or alkaline-earth cation. Their crystal structures and thermodynamic stability are governed by the structural arrangement of the [BH4] units, yielding to rich phase diagrams especially for the alkaline-earth compounds. For instance in Mg(BH4)2, the observed polymorphs are almost degenerate in their ground state energy, yielding quite often to phase mixtures during preparation, or metastable states.6–8 α-Mg(BH4)2 is energetically most favourable at low temperatures and it can be obtained as a solvent free material from solution employing a specific drying procedure.4 Upon heating to 490 K, the material undergoes a phase transition to the high temperature β-phase which then remains metastable during cooling down to, at least, 10 K.4,9 Further polymorphs of Mg(BH4)2 include a porous γ-phase and a high pressure δ-phase.9,10 Similarly, for Ca(BH4)2, there are four known crystal structures: α-, α′ and γ-phase are considered low temperature phases, while β is a high temperature phase.11–13 The hydrogen release properties differ slightly for the various polymorphs.14 The alkaline borohydrides, LiBH4, NaBH4 and KBH4 also show different crystal structures, but in contrast to their alkaline-earth relatives, the observed structural phase transitions between low and high temperature crystal structures are fully reversible as the temperature is increased above or lowered below the respective transition temperatures. (LiBH4: T = 381 K,15 NaBH4: T = 190 K16 and KBH4: T ≈ 70 K17).
The complexity and the number of different crystal modifications observed for Mg(BH4)2 is thought to be the result of the interplay between cation size and interaction between neighbouring [BH4]− anions.18 In ionic crystals, the coordination number around the central cation is given by the ratio of the size of the metal cation in its appropriate oxidation state and the size of the anion. In the borohydrides, additionally, the orientation of the tetrahedral [BH4]− anion, and the mutual repulsion of the hydrogen atoms (each carrying a negative partial charge) come into play. Energetically, a tridendentate orientation of the [BH4]− towards the cation is most favourable, most unfavourable is the monodentate orientation which has not been found in any experimental crystal structure so far.18 For Mg2+, the ion radius yields a coordination number on the border between tetragonal and octagonal coordinations, and additionally, the tetrahedral [BH4] can not be packed in a space filling manner. In combination with the various possibilities for the [BH4] orientation, this yields to a variety of predicted and experimentally observed crystal phases which are almost degenerate in energy.6–8,18 This effect is supposed to be less pronounced in the alkaline borohydrides due to the lower cation charge.
From the above, it follows that the orientation of the [BH4] units and their dynamics at non-zero temperatures have a significant impact on the stability of the compounds. In the alkaline borohydrides LiBH4, NaBH4 and KBH4, the crystal phase transition is accompanied by a change of the local rotational reorientation of the [BH4] (order–disorder transition), moreover changes in the external [BH4]-vibrational modes have been measured using inelastic neutron spectroscopy (INS).19–22 INS has also been used to measure the low energy excitation spectra of α- and β-Ca(BH4)2, showing that the two polymorphs differ in the vibrational frequency of the [BH4] tetrahedra.23 Measurements of the low energy transfer dynamics in these compounds are thus essential to understand the stability and binding in the borohydride compounds.
In the same context, multiple attempts to destabilize (or stabilize) borohydrides by the synthesis of dual cation compounds have been undertaken, i.e. stabilizing crystal structures that comprise two different cations balancing the negative [BH4]− anions. Nakamori et al.24 showed in first principles calculations that the thermodynamic stability of alkaline and alkaline-earth borohydrides correlates with the electronegativity of the cations. The use of dual cation compounds with an average intermediate electronegativity thus could offer a pathway to tune the hydrogen desorption properties to conditions more viable for applications. Dual cation compounds have been successfully synthesized and their structures were characterized for a series of Li–Zn and Na–Zn borohydrides25 or M–Sc borohydrides (with M = Li, Na, and K), for an overview see ref. 26. Moreover, mixed borohydride with KBH4 (Li–K-borohydride,27 or Mg–K-(BH4)x28) have been identified. However, for LiBH4 + Mg(BH4)2 mixtures there are contradicting findings, and both, the physical mixture of powders has been reported29,30 as well as a dual cation compound.31,32
In this paper, we will report quasielastic (QENS) and inelastic neutron spectroscopy (INS) data to elucidate the low lying excitations and local reorientations in the alkali borohydride LiBH4, the alkaline-earth borohydrides α- and β-Mg(BH4)2, and an 1:1 molar mixture LiBH4 + α-Mg(BH4)2. All samples are investigated as a function of temperature and the dynamic properties for the various crystal modifications were thus explored. LiBH4 has already been intensely studied using QENS20,33–35 or INS36 and we will briefly summarize our results as the baseline for discussion of the results for the 1:1 molar mixture LiBH4 + α-Mg(BH4)2 to further investigate the possibility of a dual cation borohydride.
At low temperature (LT-), the crystal structure of LiBH4 is orthorhombic (Pnma), and around 381 K, a first order structural phase transition occurs to the hexagonal high temperature (HT-) phase (P63mc).37 After some controversy, it has been concluded that the hydrogen atoms are bonded to the central boron in an almost perfect tetrahedral shape in both crystal modifications.15 From synchrotron diffraction experiments, the distance between B and H (dB–H) was estimated to be 1.16–1.26 Å in the HT-phase,15 whereas neutron diffraction experiments show dB–H = 1.18–1.20 Å at 302 K.38 The cell parameters of the LT-crystal structure display a highly anisotropic temperature dependence and it was suggested that the structural phase transition is driven by anharmonic effects.15 The structural phase transition is accompanied by an order–disorder transition of the [BH4] units.19,20,38
The α-modification of Mg(BH4)2 belongs to the P6122 symmetry space group with six non-equivalent positions of the BH4 tetrahedra.39 The β-phase has an orthorhombic Fddd structure in which five non-equivalent [BH4] positions have been identified. The unit cell volume is almost the double of the α-polymorph. Both α- and β-Mg(BH4)2 show anomalous thermal expansion, much more pronounced in the latter polymorph.39 In both crystal modifications, Mg atoms are surrounded by four [BH4] tetrahedra leading to a strongly distorted tetrahedral environment. At a local level, each Mg atom is facing two hydrogens of the adjacent [BH4] in a bidentate orientation, leading to an almost linear Mg–H2–B–H2–Mg chain, with two chains at 90° at each Mg-atom. The resulting MgH8 polygons form different membered rings in the respective crystal structures. The dynamics of the [BH4]− anions has been studied by Nuclear Magnetic Resonance (NMR), Raman, infrared and inelastic neutron spectroscopy. NMR measurements on β-Mg(BH4)2 show that the rotational dynamics of the [BH4] units is characterized by a broad distribution of activation energies, whereas, in α-Mg(BH4)2, NMR measurements show three distinct jump processes, with significantly higher activation energies.40–42 The difference has been attributed to the different crystal structures and to the [BH4] inequivalent crystallographic positions.
To our knowledge, so far there have been no measurements of the dynamic properties of an 1:1 molar mixture of LiBH4 + α-Mg(BH4)2. The 1:1 ratio is close to the eutectic composition of the binary system with a melting point between 450 and 520 K.30
2 Experimental
Neutron scattering measurements were performed at the cold direct geometry time-of-flight spectrometer TOFTOF operated by the Technische Universität München at the Heinz Maier-Leibnitz Zentrum (MLZ) in Garching, Germany.43,44 All measurements were performed in transmission mode, i.e. flat sample cells were oriented at 2θ = 135° with respect to the incoming beam. Data were corrected by subtracting the empty container contribution, accounting for the self-shielding factors, and normalization to a vanadium standard. For the analysis, the aluminium Bragg peaks arising from the sample environment and the sample cells were excluded. The obtained dynamic structure factor S(Q,ω) was binned into a regular grid in energy (ħω) and momentum transfer (Q). Due to the high incoherent scattering cross section of hydrogen, the signal registered at the detector is predominantly from hydrogen. Three different incident wavelengths were chosen to cover a large dynamic range. The settings were as follows (with δEi the FWHM of the resolution function at zero energy transfer):
(1) λ1 = 6 Å, with δE1 = 48 μeV and ΔQ1 = 0.2–1.8 Å−1,
(2) λ2 = 3.5 Å, with δE2 = 200 μeV and ΔQ2 = 0.3–3 Å−1.
(3) λ3 = 2.5 Å, with δE3 = 480 μeV and ΔQ3 = 0.5–4.2 Å−1.
LiBH4 was purchased from Alpha Aesar, with 95% nominal purity. The material was used without further purification. Approximatively, 70 mg of material was employed for the measurements, yielding to an estimated neutron transmission of ∼85%. Measurements were performed with the incident neutron wavelength λ3, and at 343, 373 and 403 K. Mg(BH4)2 was prepared in the α- and β-crystal modifications as described in ref. 4. In both polymorphs, the isotope 11B was used in order to minimize the neutron absorption. Approximatively, 200 mg of Mg(11BH4)2 were used, yielding to an estimated neutron transmission of ∼90%. Measurements were performed at λ1 and λ3, in the temperature range between 11 K and 500 K. Each temperature was measured for 3 hours, with additional 30 minutes of equilibration time. The data obtained for β-Mg(BH4)2 have been reported previously45 and the results will be discussed in conjunction with the data presented here.
A batch of 1:1 molar mixture of LiBH4 and α-Mg(BH4)2 was prepared by high energy ball milling according to the procedure described ref. 30. In the mixture, the isotope 11B was used in both lithium and magnesium borohydride. In total, approx. 160 mg of material were used for the measurements. Measurements were performed at λ2 and at 373, 423 and 500 K, i.e. below and above the crystal phase transitions of LiBH4 and Mg(BH4)2.
3 Data analysis
Generally, in the incoherent approximation, the scattering function S(Q,ω) for particles performing simultaneous but independent motions can be described as:| | | S(Q,ω) = Strans(Q,ω) ⊗ Srot(Q,ω) ⊗ Svib(Q,ω), | (1) |
i.e. it is given by the convolution of the scattering functions for translational diffusion, rotation and vibration, respectively. In borohydrides, in the temperature range studied here, relevant for the analysis are jump rotational diffusion and vibration processes. The scattering function of rotational motions is given by the sum of a δ-function and a series of Lorentzian functions:| | 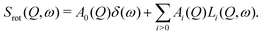 | (2) |
The Lorentzians Li(Q,ω) are characterized by their half-width at half maximum (HWHM) Γi, which are independent of the momentum transfer Q and related to the mean residence time between subsequent jumps, Γi ∼ ħτi−1. Because of the normalization condition of the scattering function, it is 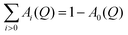 . A0(Q) is the elastic incoherent structure factor of the rotation which depends on the spatial extension and its symmetry. For simple rotations, expressions for A0(Q) can be obtained analytically, see for example ref. 46.
. A0(Q) is the elastic incoherent structure factor of the rotation which depends on the spatial extension and its symmetry. For simple rotations, expressions for A0(Q) can be obtained analytically, see for example ref. 46.
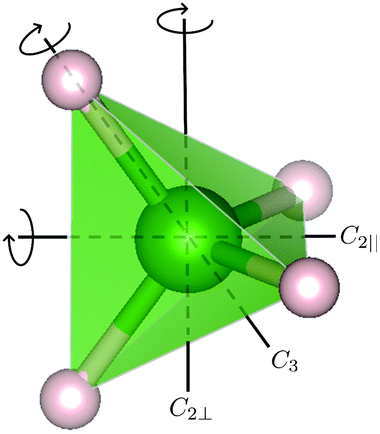
For the [BH4] tetrahedra, the most likely rotational motions occur around a C2 or C3-symmetry axis of the tetrahedra (see Fig. 1), A0(Q) for both cases is given by
| | 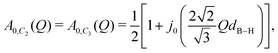 | (3) |
dB–H is the boron–hydrogen distance and
j0(
x) is the zero order Bessel function,
j0(
x) = sin
x/
x. In the case there is no preferred
C2 or
C3-axis and all four hydrogen atoms exchange places with equal probability (
C4 tumbling motion),
A0(
Q) reads:
47| | 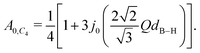 | (4) |
Vibrational motions are generally described by
| | | Svib(Q,ω) = D(Q)δ(ω) + [1 − D(Q)]Sinelvib(Q,ω), | (5) |
where
D(
Q) denotes the Debye–Waller Factor (DWF). In the harmonic and isotropic approximation, it is given by
D(
Q) = exp(−〈
u2〉
Q2/3), where 〈
u2〉 is the mean square displacement of the hydrogen atoms. In the one-phonon harmonic approximation, the inelastic scattering function
Sinelvib(
Q,
ω) can be described by a damped harmonic oscillator (DHO).
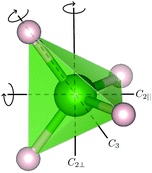 |
| | Fig. 1 Structure of the [BH4] tetrahedra with three rotation axes: 3-fold 120° rotation axis C3, and the 2-fold 180° rotation axes (C2⊥ and C2‖). (green sphere) boron, and (small grey spheres) hydrogen. | |
It has been shown previously45 that in β-Mg(BH4)2, low energy vibrational modes inhibit the separation of rotation and vibration parts of the QENS spectra. Therefore, for the analysis, the convolution of Srot and Svib had to be considered in full. The measured S(Q,ω) was described using:
| S(Q,ω) ∼ Rres(ω) ⊗ [EISF·δ(ω) + QISFL1·L1(Γ,Q) |
| | | + QISFvib·DHO(Q,ωq,ΓDHO)] + bg(Q), | (6) |
where the prefactors EISF, QISF
L1 and QISF
vib give the partial contribution of the elastic, quasielastic and inelastic components of the scattered intensity.
Rres(
ω) is the (measured) resolution function of the instrument and the linear background bg(
Q) takes motions into account that are outside the observation time window of the experiment. Details on the derivation of the fit function can be found in the ESI.
† As a result of the fit, the structure factors EISF, QISF
L1 and QISF
vib are obtained, as well as the HWHM
Γ1 proportional to the time scale of the jump rotation. The factors EISF, QISF
L1 and QISF
vib depend on the rotational elastic incoherent structure factor,
A0(
Q), the fraction of activated rotations, 1 −
p and the Debye–Waller Factor,
D(
Q) as follows:
| | | EISF = D(Q)[p + (1 − p)A0(Q)] | (7) |
| | | QISFL1 = D(Q)(1 − p)[1 − A0(Q)] | (8) |
A0(
Q),
p and
D(
Q) are thus obtained from a simultaneous fit of the prefactors to
eqn (7)–(9). The equations above are valid assuming only a single rotation with characteristic jump frequency
τ−1 ∼
Γ/
ħ. The extension for two characteristic rotation modes having distinct time scales is given in the ESI.
†
4 Results and discussion
4.1 α-Mg(BH4)2
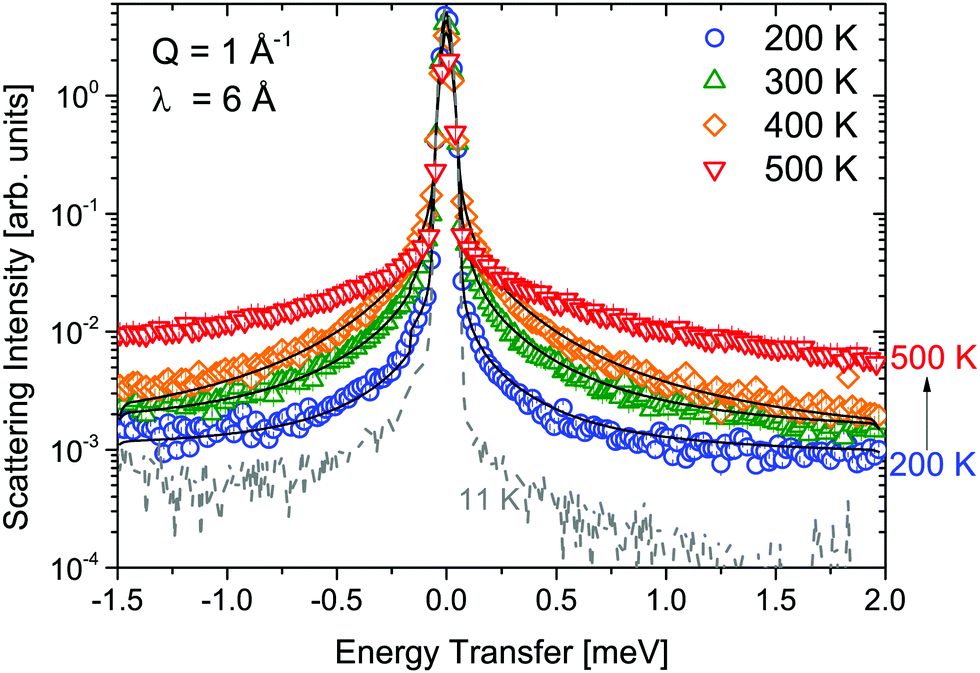
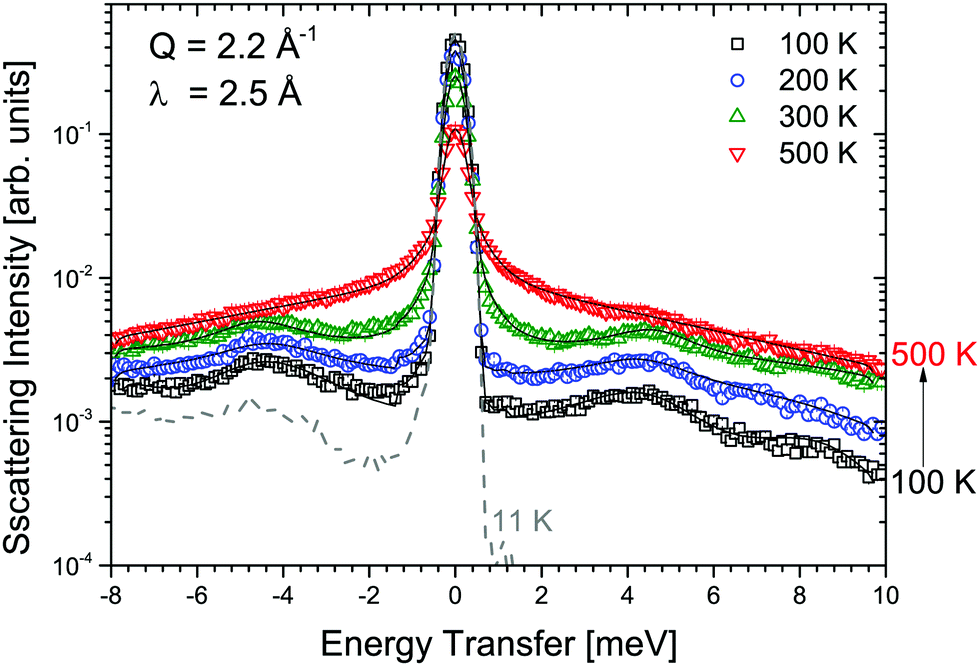
Measurements of α-Mg(BH4)2 were performed at λ1 = 6 Å and λ3 = 2.5 Å and the obtained S(Q,ω) are shown in Fig. 2 and 3, respectively. Measurements were taken at temperatures from 11 to 500 K. Above 490 K, α-Mg(BH4)2 transforms into the β-phase. The measurements at 6 Å (probing timescales up to ∼16 ps) show a quasielastic broadening already at 200 K, and its contribution increases with increasing temperature. When shorter time scales are probed (2.5 Å-data, timescales up to ∼1.6 ps), the spectra are dominated by inelastic excitations that are observed in addition to the weak quasielastic signal around the elastic line. With the phase transition to the β-phase, both the quasielastic and inelastic signals undergo a distinct transformation and the data measured at 500 K were consistent with the findings published for β-Mg(BH4)2.45 The low lying excitations in α-Mg(BH4)2 have not been reported in previous studies of hydrogen dynamics in the α-phase.48,49 The data analysis was conducted as described in the previous section. Both data sets could be fitted using a combination of quasielastic and inelastic components. The results of the fits are included in Fig. 2 and 3 (solid lines). The measurements at longer time scales (6 Å data) indicate that there are two quasielastic processes, with distinct Γ1,α and Γ2,α (both independent of Q) in addition to the vibrational part (modeled as a flat background). The values of Γ1,α and Γ2,α obtained for the quasielastic components are given in Table 1.
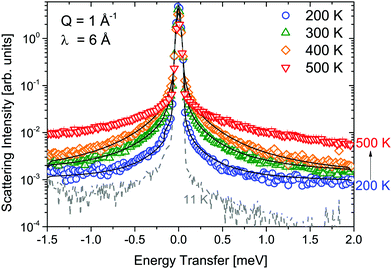 |
| | Fig. 2 α-Mg(BH4)2: S(Q,ω) measured at λ1 = 6 Å and at 11, 200, 300, 400 and 500 K. The solid black lines represent the fit to the data. The vertical error bars denote ±1σ. | |
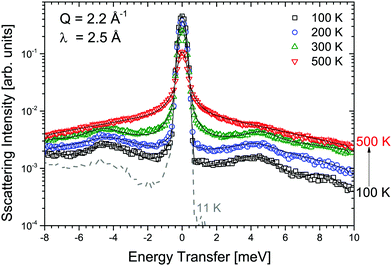 |
| | Fig. 3 α-Mg(BH4)2: S(Q,ω) measured at λ3 = 2.5 Å and at 11, 100, 200, 300 and 500 K. The solid black lines represent the fit to the data. Gray dashed line: 11 K (resolution), black squares: 100 K, blue circle: 200 K, green up triangles: 300 K, and red down triangles: 500 K. Data measured at 400 K have been omitted for clarity. The vertical error bars denote ±1σ. | |
Table 1 Widths Γ1,α and Γ2,α obtained from the fits of the measurements of α-Mg(BH4)2 at 6 Å. No quasielastic signal was observed at 100 K. On the right side of the table the corresponding values Γi,β for β-Mg(BH4)2 are listed, data taken from ref. 45
|
T [K] |
Γ
1,α [μeV] |
Γ
2,α [μeV] |
Γ
1,β [μeV] |
Γ
2,β [μeV] |
|
At 500 K, both samples are in the β-modification.
|
| 100 |
— |
— |
— |
— |
| 200 |
30 ± 60 |
280 ± 150 |
33 ± 2 |
330 ± 50 |
| 300 |
26 ± 30 |
300 ± 60 |
73 ± 11 |
350 ± 40 |
| 400 |
39 ± 3 |
300 ± 50 |
80 ± 8 |
500 ± 60 |
| 500 |
100 ± 20a |
670 ± 99a |
110 ± 20 |
550 ± 80 |
Surprisingly, Γ1,α and Γ2,α were, within the experimental uncertainty, independent of temperature. At 500 K, the β-modification is reached and both widths showed a steep increase, indicative for the quickening of the motions. In comparison, the metastable β-phase exhibited slightly faster motions and a larger temperature dependence at temperatures below 500 K. Similarly to the α-phase, two rotational motions were identified and the values Γi,β are included in Table 1 (values taken from ref. 45). Table 2 shows the mean resident times of H atoms between two successive jumps around the C2 rotation axis, together with the β-Mg(BH4)2 data in the same temperature range.
Table 2 Mean resident times τi,α|β of H atoms between rotational jumps. τi,α|β = 2ħ/Γi,α|β for a C2 rotational model. In the case of C3 jump rotations τi,α|β = 3ħ/2Γi,α|β. On the right side of the table, the corresponding values for β-Mg(BH4)2 are listed, data taken from ref. 45
|
T [K] |
τ
1,α [ps] |
τ
2,α [ps] |
τ
1,β [ps] |
τ
2,β [ps] |
|
At 500 K, both samples are in the β-modification.
|
| 100 |
— |
— |
— |
— |
| 200 |
43 ± 90 |
5 ± 3 |
40 ± 2 |
4.0 ± 0.6 |
| 300 |
51 ± 58 |
4.3 ± 0.8 |
18 ± 3 |
3.7 ± 0.4 |
| 400 |
33 ± 2 |
4.4 ± 0.7 |
16 ± 2 |
2.6 ± 0.3 |
| 500 |
13.2 ± 3.0a |
1.9 ± 0.3a |
12 ± 2 |
2.4 ± 0.3 |
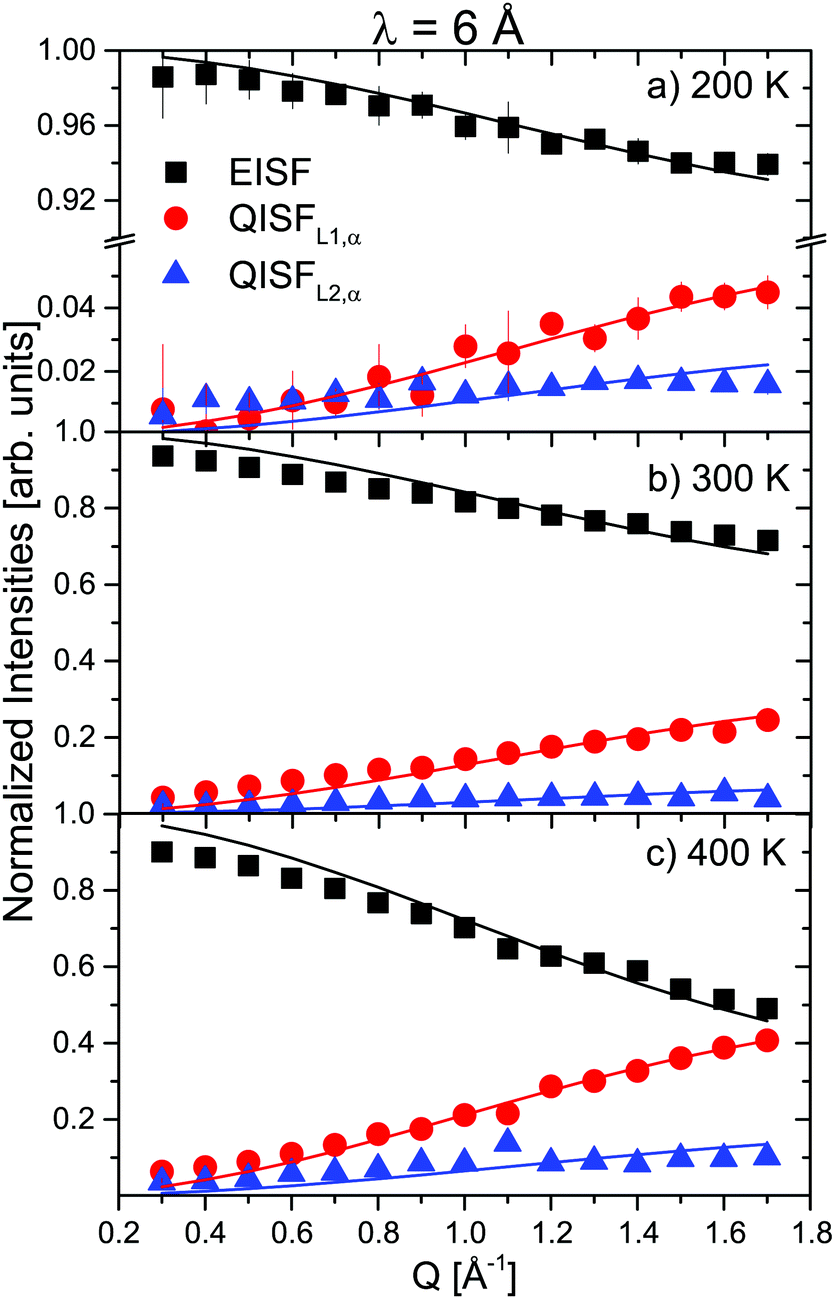
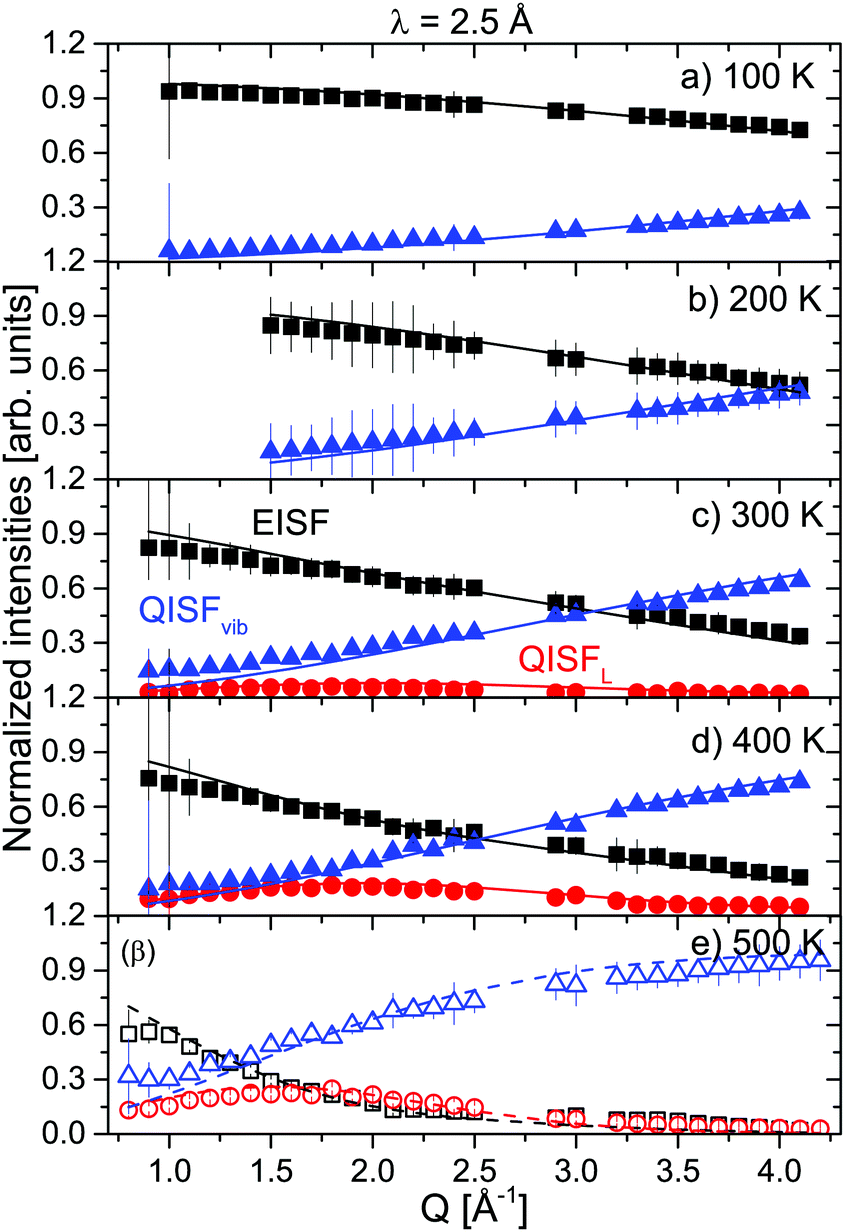
The prefactors EISF, QISFL1,α and QISFL2,α obtained from the fits to S(Q,ω) are shown in Fig. 4 together with a simultaneous fit to eqn (S14)–(S16), in the ESI.† The result indicated that both rotational motions are hindered at low temperatures, and the data were consistent with rotations around either the C2 or C3 symmetry axes of the tetrahedra. The data measured at 2.5 Å were mainly characterized by the presence of inelastic excitations at energy transfers ΔE1 ∼ ±4.2 meV and ΔE2 ∼ 8 meV. The excitations did not show any dispersion, and their position did not change with increasing temperature. A quasielastic signal could be distinguished at 300 K and above, however only at 500 K, it became considerably larger compared to the inelastic parts, i.e. only after the sample has transformed to the β-phase. From the 6 Å measurements, it was clear that the onset of the rotational motions is around 200 K, however, the expected width was too close to the energy resolution and the signal was too weak to discern at low temperatures in the 2.5 Å measurements. The narrower of the two Lorentzians was hidden in the experimental resolution at all measured temperatures. Therefore, the analysis of the 2.5 Å data below 500 K shows two DHOs and one quasielastic component in addition to the elastic line. The results of the fits are plotted in Fig. 3 (solid lines), and the structure factors EISF and QISF and QISFvib are shown in Fig. 5.
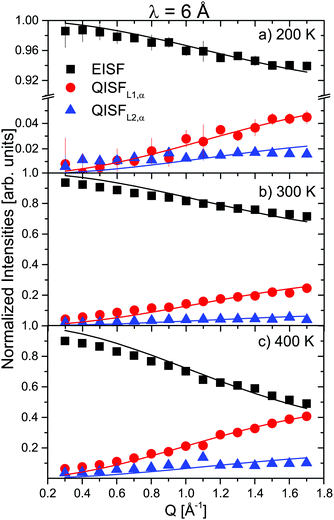 |
| | Fig. 4 α-Mg(BH4)2: EISF (black squares), QISFL1,α (red circles) and QISFL2,α (blue triangles) at different temperatures (6 data). Solid lines represent fit to the data according to eqn (S14)–(S16), in the ESI.† The vertical error bars denote ±1σ. | |
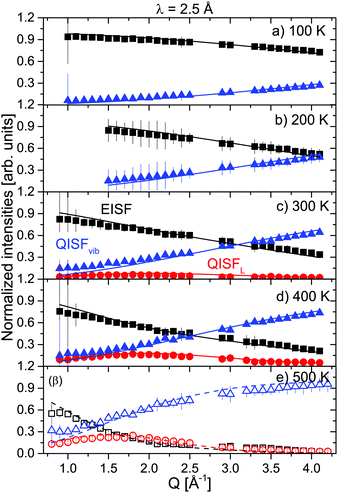 |
| | Fig. 5 α-Mg(BH4)2: EISF (black squares), QISFL (red circles) and QISFvib (blue triangles) measured at 2.5 Å and at 100, 200, 300, 400 and 500 K. Solid lines represent the fit to data according to eqn (7)–(9). Panel (e) 500 K shows open symbols to distinguish between α- and β-phases. The vertical error bars denote ±1σ. | |
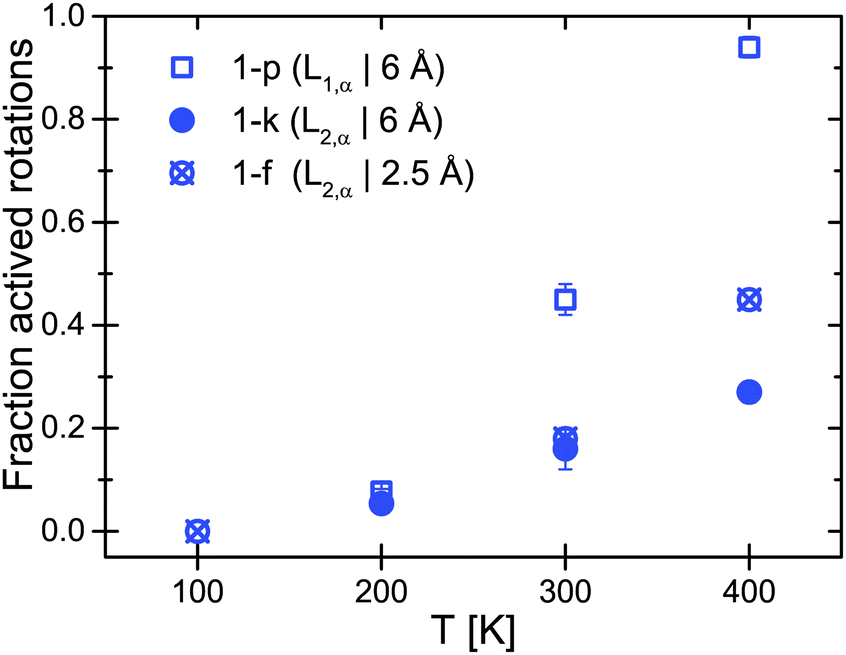
The EISF and QISFs factors were evaluated using eqn (7)–(9). In agreement with the 6 Å results, hindered rotations around the C2 or C3 axis were observed at 300 K and above, with only a limited fraction of the [BH4] tetrahedra taking part in the rotations (∼20% at 300 K and ∼40% at 400 K) (see Fig. 6). Apparently, the inequivalent [BH4] sites in α-Mg(BH4)2 have different activation energies and only in the β-modification, all rotations are activated in the observation time window.
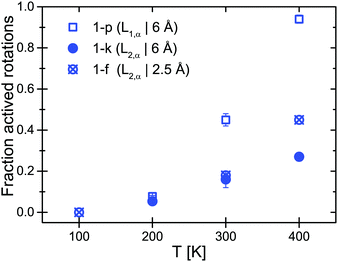 |
| | Fig. 6 α-Mg(BH4)2: fraction of the activated rotation processes (1 − p) and (1 − k) derived from EISF and QISF at 6 Å (blue open squares and solid circles) and (1 − f) obtained from the 2.5 Å measurement (crossed circles). The vertical error bars denote ±1σ. | |
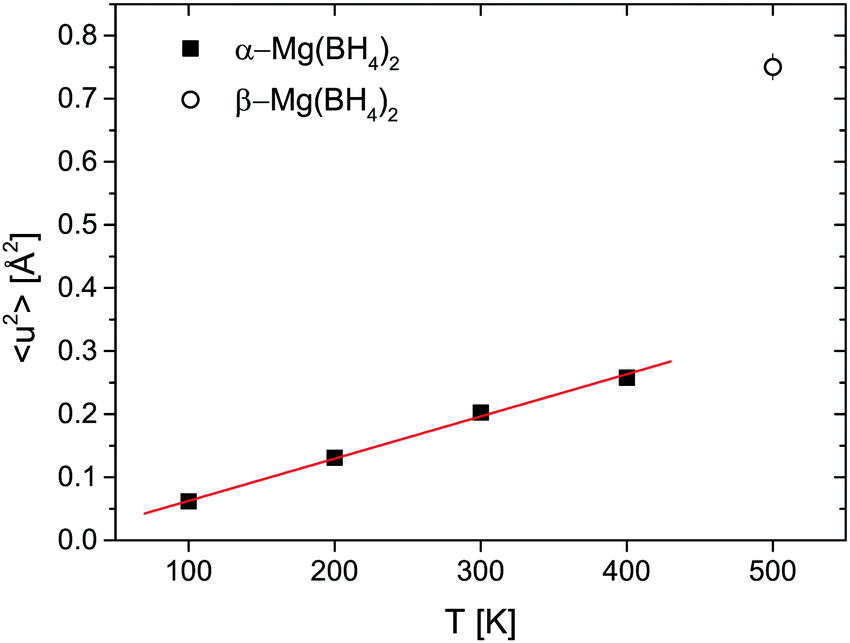
Fig. 7 shows the mean square displacement 〈u2〉 obtained from the Debye–Waller-Factor D(Q). 〈u2〉 increases linearly in the 100–400 K temperature range with 〈u2〉 = (6.7 ± 0.2) × 10−4 × T [K] Å2. The transition to the β-phase is marked by a jump-like increase of 〈u2〉.
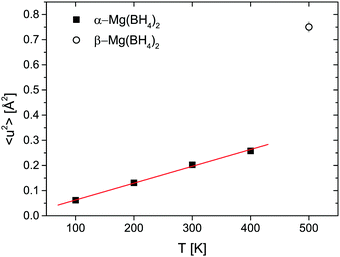 |
| | Fig. 7 α-Mg(BH4)2: mean square displacement 〈u2〉 as a function of temperature T. The red line is a linear fit to the data, in order to extract the temperature dependence: 〈u2〉 = (6.7 ± 0.2) × 10−4 × T [K] Å2. Open symbols show data at 500 K, where the system is already in the β-polymorph. The vertical error bars denote ±1σ. | |
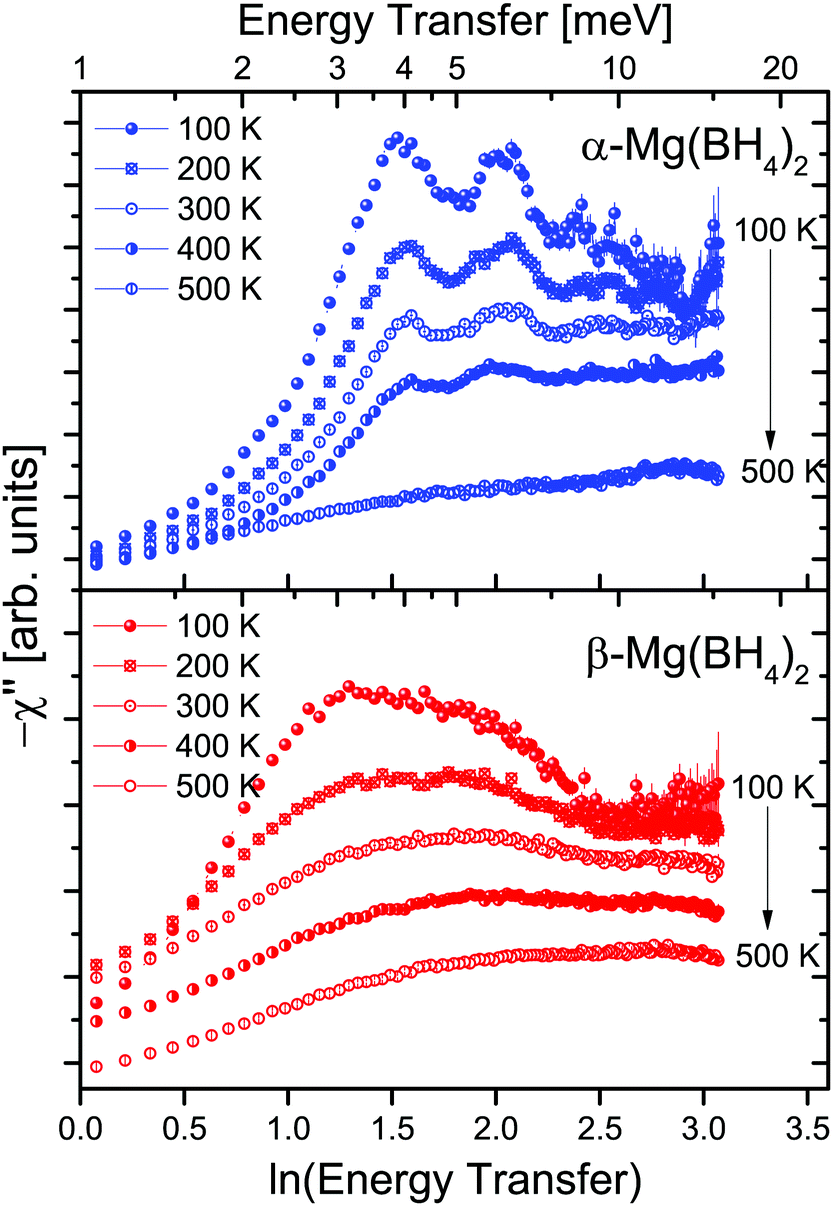
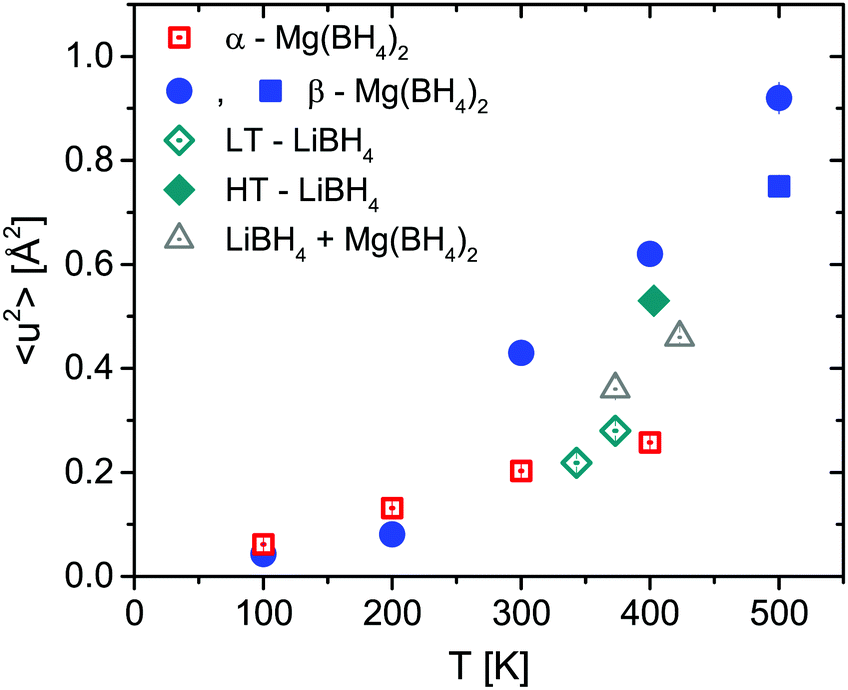
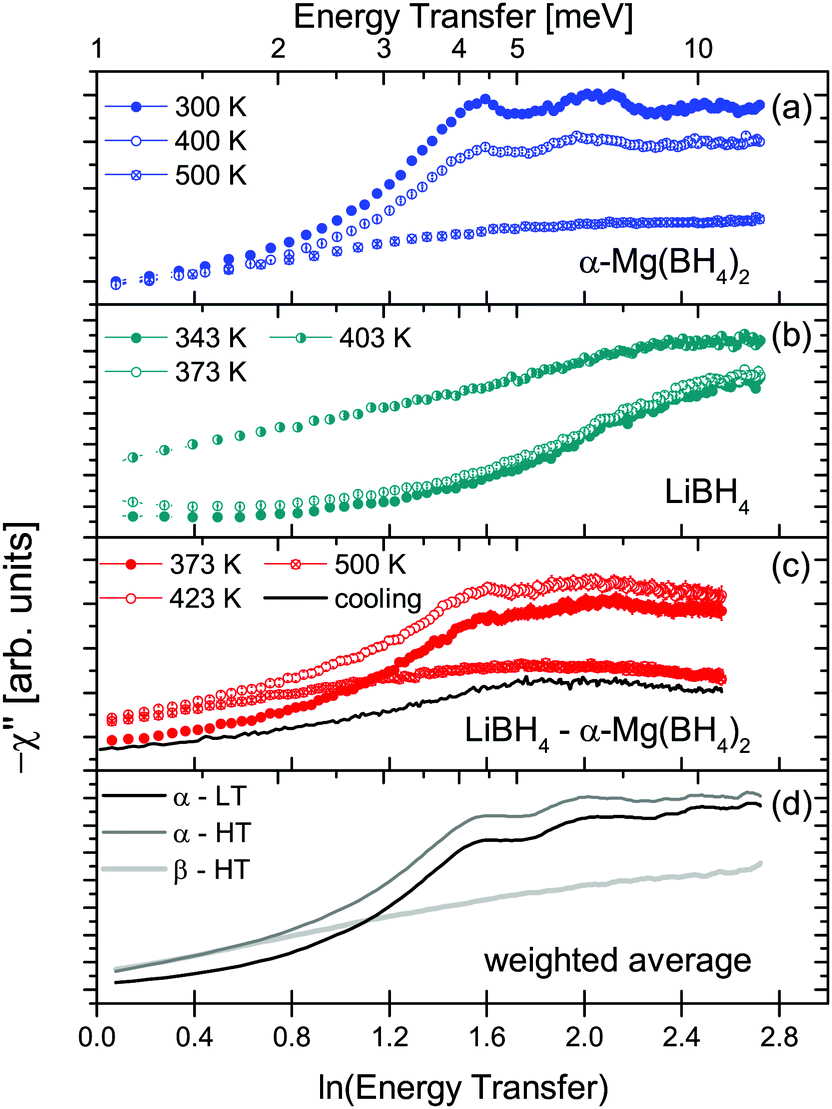
The two investigated polymorphs of Mg(BH4)2, α and β, exhibit quite different low energy dynamics, as shown above. Therefore, the analysis was mainly focused on the quasielastic scattering contribution and the local reorientation. To focus on the low energy excitation spectrum, a plot of the imaginary part of the dynamic susceptibility, χ′′ is shown in Fig. 8. The latter was calculated from 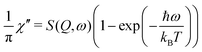 , with kB the Boltzmann constant. Data were summed over Q in the 1.5 ≤ Q ≤ 4.25 Å−1 range. For α-Mg(BH4)2, distinct peaks at around 4.2 and 8 meV are visible which hardly change with temperature. In contrast, the β-polymorph has slightly softer low energy excitations and the features are much broader and less defined. The plot underlines that β-Mg(BH4)2 shows more dynamical disorder compared to the α-phase, starting already at temperatures as low as 200 K. This is also reflected in the mean square displacement 〈u2〉, as illustrated in Fig. 9. The mean square displacement of α-Mg(BH4)2 follows a linear relationship with increasing temperature. In the β-modification, 〈u2〉 follows approximately the same relationship below 200 K, but between 200 and 300 K, a jump like increase is observed. In the same temperature region, the jump rotation processes in β-Mg(BH4)2 become fully activated45 whereas the rotation modes in α-Mg(BH4)2 are still significantly hindered even at 400 K. Incidentally, the anomalous lattice expansion observed in β-Mg(BH4)2 shows a change from contraction to the expansion of the a-lattice parameter around 200 K and the opposite for the b-axis lattice parameter, indicating a link between the local rotational dynamics, lattice dynamics and structure. In α-Mg(BH4)2, anomalous expansion is much smaller and it is restricted to temperatures below 200 K.39
, with kB the Boltzmann constant. Data were summed over Q in the 1.5 ≤ Q ≤ 4.25 Å−1 range. For α-Mg(BH4)2, distinct peaks at around 4.2 and 8 meV are visible which hardly change with temperature. In contrast, the β-polymorph has slightly softer low energy excitations and the features are much broader and less defined. The plot underlines that β-Mg(BH4)2 shows more dynamical disorder compared to the α-phase, starting already at temperatures as low as 200 K. This is also reflected in the mean square displacement 〈u2〉, as illustrated in Fig. 9. The mean square displacement of α-Mg(BH4)2 follows a linear relationship with increasing temperature. In the β-modification, 〈u2〉 follows approximately the same relationship below 200 K, but between 200 and 300 K, a jump like increase is observed. In the same temperature region, the jump rotation processes in β-Mg(BH4)2 become fully activated45 whereas the rotation modes in α-Mg(BH4)2 are still significantly hindered even at 400 K. Incidentally, the anomalous lattice expansion observed in β-Mg(BH4)2 shows a change from contraction to the expansion of the a-lattice parameter around 200 K and the opposite for the b-axis lattice parameter, indicating a link between the local rotational dynamics, lattice dynamics and structure. In α-Mg(BH4)2, anomalous expansion is much smaller and it is restricted to temperatures below 200 K.39
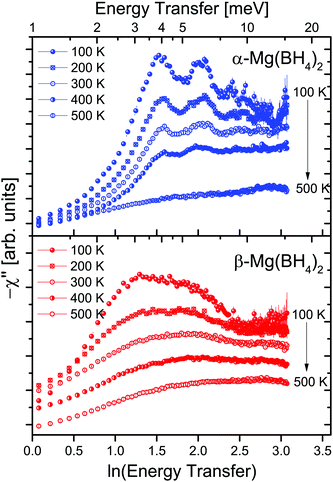 |
| | Fig. 8 −χ′′ as a function of ln (energy transfer) and at different temperatures (100, 200, 300, 400 and 500 K). α-Mg(BH4)2 shows distinct peaks up to high temperatures while β-Mg(BH4)2 has a much broader and slightly softer low energy spectrum. Data are summed for Q = 1.5–4.25 Å−1. The increase towards low energy transfers is due to the quasielastic contributions. The vertical error bars denote ±1σ. | |
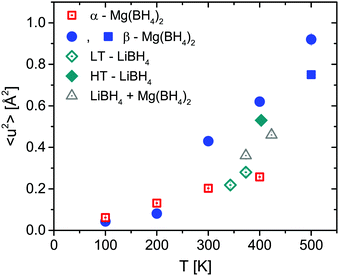 |
| | Fig. 9 Mean square displacement 〈u2〉 as a function of temperature for LiBH4, α- and β-Mg(BH4)2 and the mixture. The vertical error bars denote ±1σ. | |
The jump frequencies of the rotations in α-Mg(BH4)2 appeared to be almost independent of temperature (see Table 1), however, keeping in mind that Γi originate from hindered rotation modes and thus are an average over different sites. Usually, the temperature dependence of the jump frequency is used to estimate the apparent activation energy Ea of the motion, assuming an Arrhenius type behaviour. In the case of α-Mg(BH4)2, the temperature independent width of the Γi would yield to a unrealistically low value of Ea. Apparently, the differences in two crystal structures are sufficient to effectively lock the [BH4] tetrahedra in position in the α-phase while enabling more degrees of freedom in the β-phase. These differences must have their origin in the arrangement of the Mg–[BH4] coordination polyhedra. The low energies probed in the current experiments are characteristic for acoustic and optical modes within the Mg–[BH4] framework7 while the (pure) vibrational frequencies of the [BH4] units have been reported to be around approximately 65 meV.48,49 The internal bending and stretching modes of the [BH4] tetrahedra are at even higher energies and IR and Raman spectroscopy data show that the bending and stretching modes of the [BH4] are quite similar for the α- and β-modifications.9 However, the long range arrangement of the B–H2–Mg–H2–B-units and the resulting 3D network is different for the two polymorphs.39 In particular, the range of Mg–H distances observed in β-Mg(BH4)2 is slightly larger compared to the α-phase (β: dMg–H = 1.90–2.19 Å, α: 1.92–2.16 Å) and the intermolecular H⋯H distance between adjacent [BH4] units is in average shorter in the α-phase.9,50 Additionally, the average distance between B and Mg is larger in the β-polymorph compared to the α-phase (β: dMg–B = 2.34–2.49 Å and α: 2.39–2.45 Å). Although both polymorphs show a chain-like structure and the same Mg–[BH4]–Mg local environment, the different crystallographic distances between B, Mg, and H are at the origin of the different vibrational and reorientational dynamics. The mutual repulsion of the negatively charged hydrogen atoms together with a covalent character of the Mg–H bond prevents the reorientation of adjacent [BH4] units and this can be at the origin of the lower mobility observed in the α-phase. On the other hand, once the material is in the β-phase, the dynamic reorientation of the [BH4] units might prevent the rearrangement of the Mg–[BH4] framework to the α-configuration down to low temperatures. [BH4] rotations remain substantially activated in the β-phase at least to 200 K, and at this point, kinetics of the first order structural phase transition has slowed down considerably. Differences in the dynamics of the [BH4] units have also been reported from NMR measurements41,42 which found higher mobility and faster rotations in β-Mg(BH4)2 compared to the α-phase. In fact, the latter shows a much higher activation energy for reorientations compared to the β-polymorph, and the rotational dynamics is one order of magnitude slower.
4.2 LiBH4
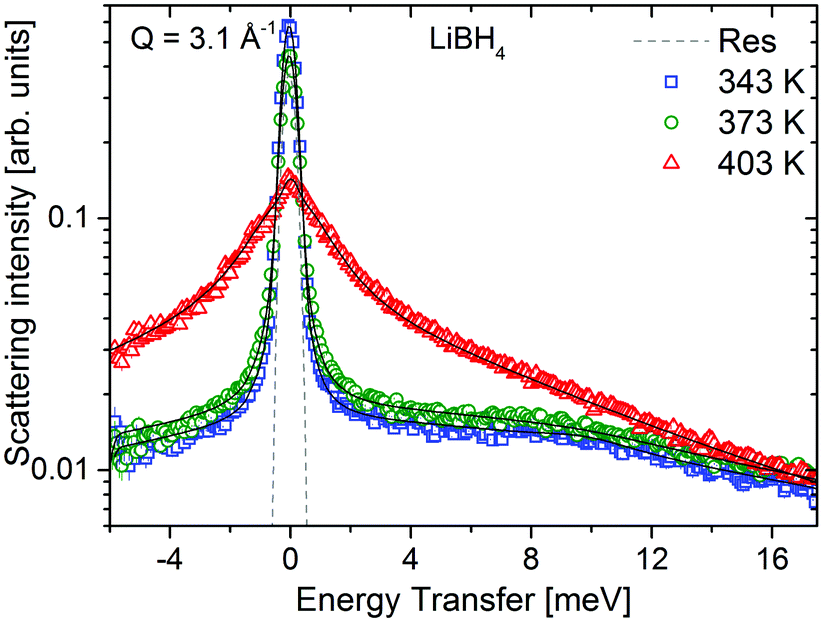
LiBH4 was measured at temperatures below and above its crystal phase transition at 381 K. Below 381 K, LiBH4 is in the hexagonal LT-modification and the neutron spectrum consists of a quasielastic component originating from jump rotations, and a vibrational part with an explicit feature centered around 9 meV. The spectra are shown in Fig. 10, measurements were taken at λ3 = 2.5 Å. Above 381 K, the sample is in the HT-modification, and the scattering intensity is dominated by a significantly broadened quasielastic contribution. These data are in agreement with previously published results on LiBH4.20,35 In contrast to Mg(BH4)2, the crystal phase transition in LiBH4 is fully reversible with temperature. The same procedure as described above was used to analyze the data. In the LT-phase, data were modeled using one quasielastic component with characteristic width, Γ1 stemming from rotations, the elastic contribution and a vibrational background. The latter was modeled either by a DHO + (broad Lorentzian) in the LT-phase or a broad Lorentzian in the HT-phase.
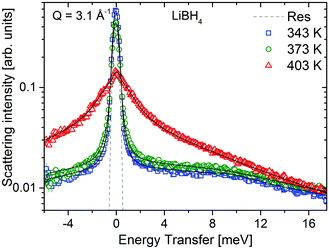 |
| | Fig. 10 LiBH4: S(Q,ω) at Q = 3.1 Å−1 and at 343 K (blue squares), 373 K (green circles) and 403 K (red triangles). The dashed line is the resolution function measured with a vanadium standard. The solid black lines represent fits to the data. The vertical error bars denote ±1σ. | |
The width, Γ1, of the quasielastic component is independent of the momentum transfer Q. The values are summarized in Table 3 and are in good agreement with previous reports on LiBH4.20,35
Table 3 HWHM Γ1 of LiBH4 in the LT- and HT-modifications. The mean resident time is calculated according to the jump rotational model at different temperatures (see the ESI)
|
T [K] |
Γ
1,LiBH4 [meV] |
τ [ps] |
| 343 |
0.14 ± 0.02 |
7 ± 1 |
| 373 |
0.21 ± 0.02 |
3.1 ± 0.3 |
| 403 |
1.43 ± 0.09 |
0.46 ± 0.03 |
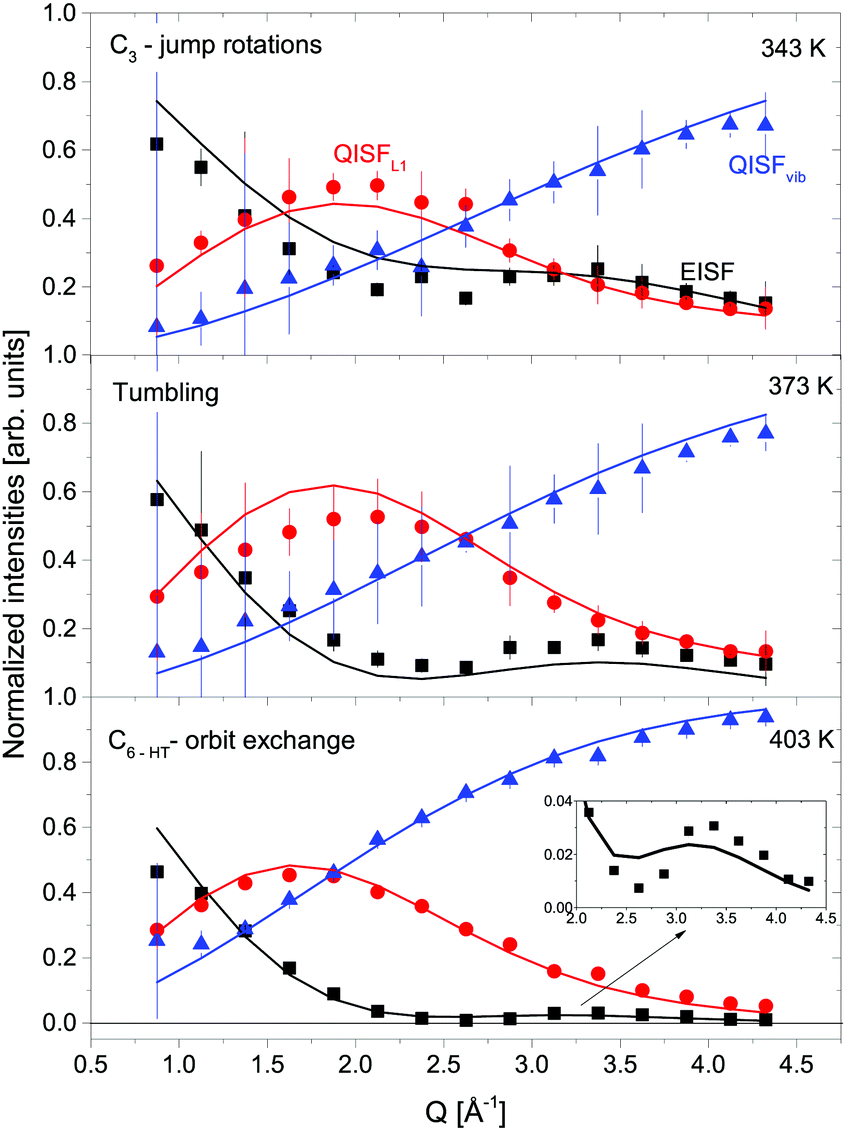
The structure factors EISF, QISFL1 and QISFvib are plotted in Fig. 11. For the evaluation, simultaneous fit using eqn (7)–(9) and A0(Q) of the previously reported rotation modes were employed. At 343 K, a rotation around the C3 axis was assumed,20 and at 403 K the orbit exchange model (where the [BH4] units rotate quasi free around one of the C3-symmetry axes with only occasional flips of that axis20), in agreement with previous results. The structure factors obtained at 373 K already appear to be somewhat “in between” the lower and higher temperature measurements, indicating that the low energy dynamics are changing below the crystal phase transition. The extracted structure factors are inconsistent with rotations solely around the C3-symmetry axis of the tetrahedra but a simultaneous fit of EISF, QISF and QISFvib assuming tumbling rotational motion yielded satisfactory results. The mean square displacements 〈u2〉 at 343 and 373 K are ∼0.2–0.3 Å2, and the value strongly increases to 0.53 ± 0.01 Å2 at 403 K. Fig. 9 shows the mean square displacement of LiBH4, together with the values obtained for α- and β-Mg(BH4)2. It should be noted that the values obtained for the LT-modification are comparable with α-Mg(BH4)2, while HT-LiBH4 resembles β-Mg(BH4)2.
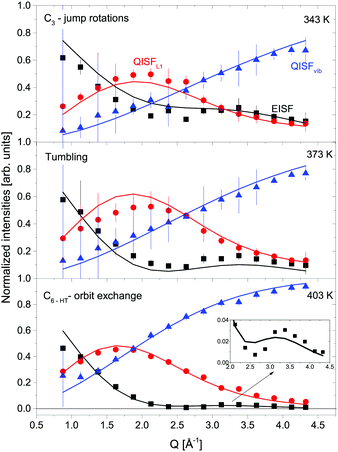 |
| | Fig. 11 LiBH4: normalized intensities at different temperatures (from top to bottom): EISF (black squares), QISFL1 (red circles) and QISFvib (blue triangles). Solid black lines represent the fit to the data according to different A0(Q), see the text for details. Vertical error bars denote ±1σ. | |
4.3 LiBH4 + α-Mg(BH4)2
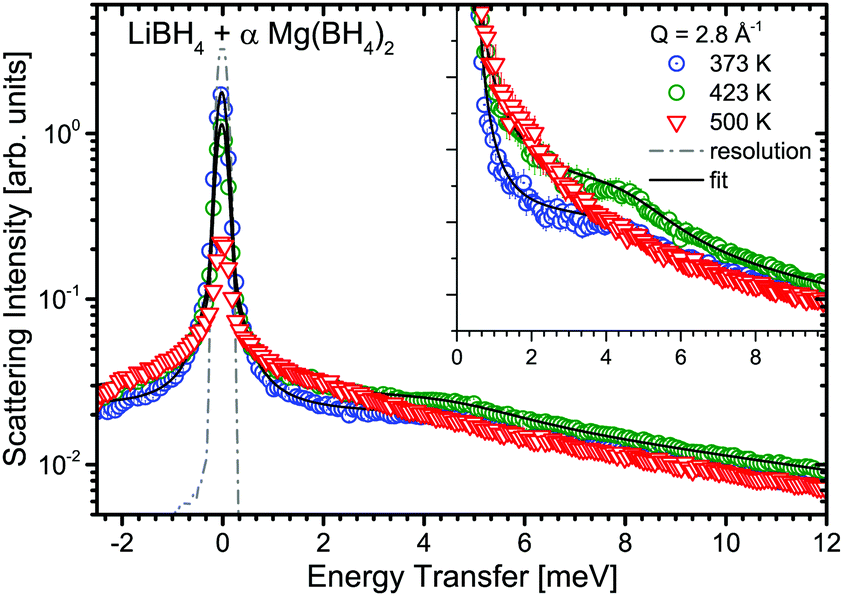
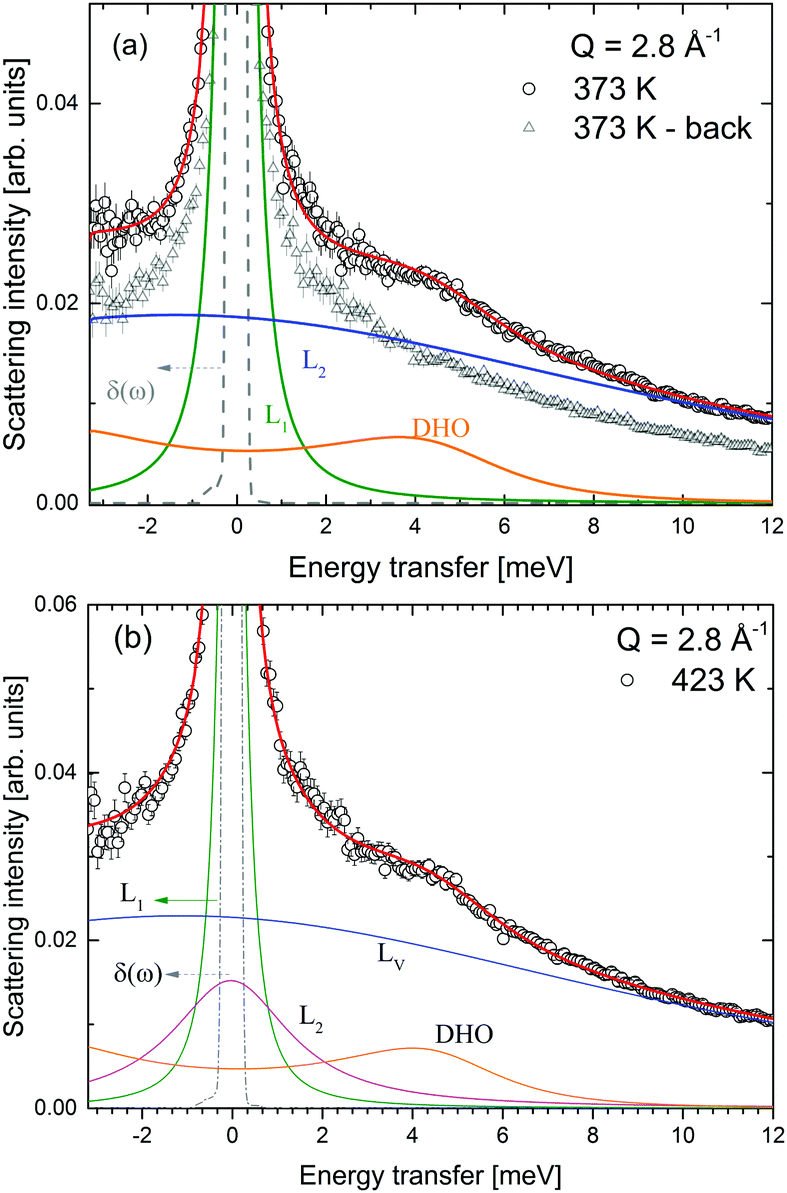
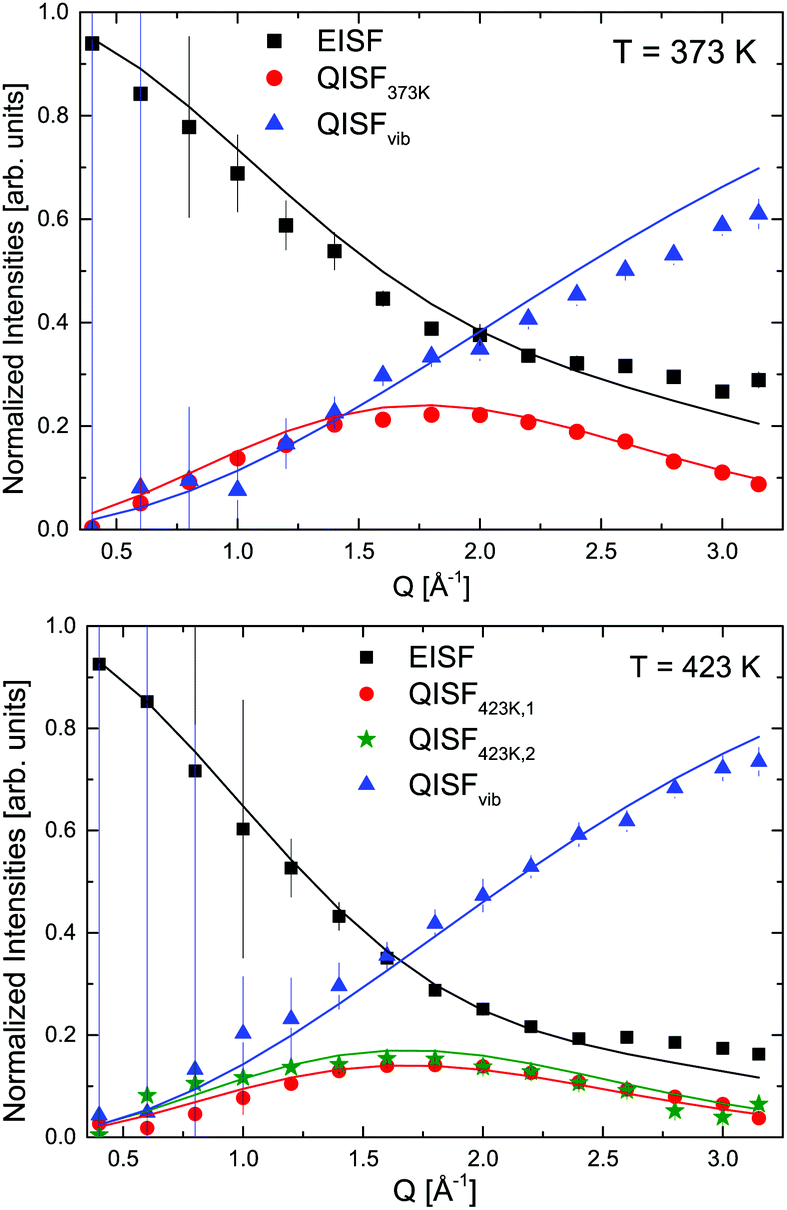
The mixture LiBH4 + α-Mg(BH4)2 (1:1 molar ratio) was measured at λ2 = 3.5 Å at temperatures between 373 and 500 K, i.e. below and above the structural phase transition of the constituents, the data are shown in Fig. 12. The slightly better energy resolution was chosen to enable a better discrimination of the narrow quasielastic components compared to measurements at λ2 = 2.5 Å. As for the pure compounds before, the spectra are composed of quasielastic and inelastic components, most prominently, the inelastic excitation of α-Mg(BH4)2 at 4.2 meV is observed. The data analysis therefore assumed a physical mixture of LiBH4 and Mg(BH4)2, each in the respective crystal modification expected at the given temperature. Since the neutron scattering signal originates almost exclusively from the hydrogen atoms in the sample, the composite spectrum can be assumed to contain 2/3 Mg(BH4)2 like features and 1/3 stemming from LiBH4. Such an ansatz yields a reasonable description of the measured S(Q,ω) for temperatures below 500 K as can be seen in Fig. 13. The graph shows the measured data together with the total fit function, as well as the split into the quasielastic and vibrational components. At 373 K, L1 describes the rotational contribution of α-Mg(BH4)2 and LT-LiBH4 (denoted Γ2,α and Γ1,LiBH4 in the above sections), however, due to the closeness of the expected width, a distinction between Mg(BH4)2 and LiBH4 could not be done. DHO and LV are attributed to the vibrational part of α-Mg(BH4)2 and LiBH4, respectively. At 423 K, LiBH4 has undergone the phase transition to the HT-phase, while Mg(BH4)2 remains in the α-modification. As can be seen in Fig. 13, a further quasielastic contribution is observed whose width Γ2 ∼ 1.6 meV is reminiscent of the quasielastic component Γ1,LiBH4 at 403 K.
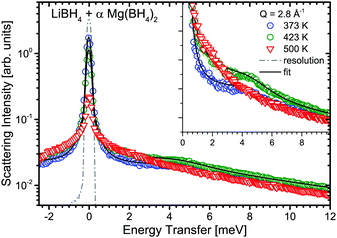 |
| | Fig. 12 LiBH4 + α-Mg(BH4)2: S(Q,ω) of an 1:1 molar mixture at 373, 423 and 500 K at Q = 2.8 Å−1. The dashed line represents the resolution function of the instrument. The inset zooms into the energy transfer region around 4 meV. Solid black lines are fits to the data. The vertical error bars denote ±1σ. | |
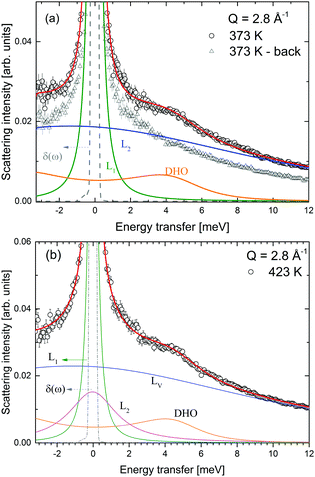 |
| | Fig. 13 LiBH4 + α-Mg(BH4)2: S(Q,ω) (open symbols), fit to the data (red solid line) and the respective contributions to the spectra at (a) 373 K and (b) 423 K. In panel (a), the data after cooling the sample from 500 to 373 K (triangles) are also shown. The vertical error bars denote ±1σ. | |
The elastic structure factors EISF, QISFi and QISFvib were obtained and the results are shown in Fig. 14. Assuming a 2/3:1/3 ratio for the Mg(BH4)2 and LiBH4 like features, the structure factors follow the set of equations (for details see the ESI†):
| | 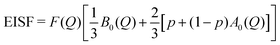 | (10) |
| | 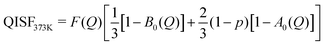 | (11) |
| | 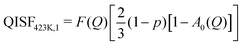 | (12) |
| | 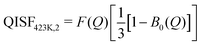 | (13) |
where the only free parameters are
F(
Q), the average Debye–Waller Factor and 1 −
p, the fraction of activated rotations in α-Mg(BH
4)
2. Depending on temperature, one or two distinct quasielastic contributions were identified and the corresponding expressions for QISF
373K and QISF
423K,i, (
i = 1, 2) were used. The rotational elastic structure factors
A0(
Q) and
B0(
Q) were chosen as for the single compounds,
i.e. A0(
Q) describing rotations around the
C2/
C3 symmetry axis in Mg(BH
4)
2 at 373 and 423 K, while
B0(
Q) describes rotations in LiBH
4 which change from tumbling motion to the orbit exchange. The results of the simultaneous fit are plotted in
Fig. 14 (solid lines). The average mean square displacements 〈
u2〉 are 0.36 ± 0.02 Å
2 and 0.46 ± 0.01 Å
2 at 373 and 423 K, while the fraction of activated rotations in Mg(BH
4)
2, 1 −
p, amounts to 0.21 ± 0.04 and 0.66 ± 0.03. These values are in very good agreement with the values found for the pure compounds and underline that during ball milling, LiBH
4 + α-Mg(BH
4)
2 remains as a physical mixture of the powders. The average 〈
u2〉 values lie between the values found for the pure compounds, with the exception of 373 K: here, the value is slightly higher than would be expected for the mixture.
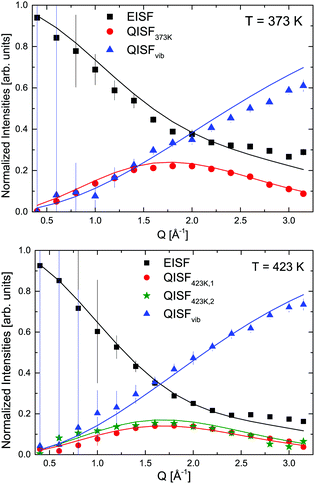 |
| | Fig. 14 LiBH4 + α-Mg(BH4)2: EISF, QISFi and QISFvib obtained from an 1:1 molar ratio for 373 and 423 K (symbols). Solid lines describe simultaneous fits to the data according to eqn (10)–(14). The vertical error bars denote ±1σ. | |
These conclusions are supported by a plot of the imaginary part of the dynamic susceptibility. Fig. 15 shows −χ′′ for the pure compounds and the mixture. In panel (d), the weighted average of α-Mg(BH4)2 and LiBH4 of −χ′′ is calculated (2/3 α-Mg(BH4)2 + 1/3 LiBH4, according to the number of hydrogen atoms in each of the constituents). Comparison with the measured data, displayed in panel (c), shows that the low energy excitation spectrum in the ball milled mixture can be well described as the weighted average of the single compounds. At 373 K, it is a mixture of α-Mg(BH4)2 and LT-LiBH4, at 423 K, it is a mixture of α-Mg(BH4)2 and HT-LiBH4, and finally, at 500 K, it is described as a superposition of β-Mg(BH4)2 and HT-LiBH4. The latter indicates that even in the melted state, the main characteristics of the individual constituents prevail, although the measured curve shows a slightly larger decrease at energies above 8 meV which is not seen in the calculated curve. It should be noted that hydrogen decomposition started at that point, and no correction for the loss in scattering intensity has been done. After cooling (to 373 K, solid line in Fig. 15(c) and data in Fig. 13(a)), the low energy spectrum is slightly changed, with a broad excitation between 5–8 meV which could be a hint towards the formation of a mixed phase. Further studies are needed to characterize the dynamics of the mixture at 500 K and after cooling. It should be noted that the melted state is accompanied by a distinct increase of the quasielastic contribution, as can be seen in Fig. 12. The elastic scattering contribution decreases significantly, however, it does not disappear completely. This is an indication that even in the melted state, there are still components that are too slow to show translatory movement in the experimental time window. However, a tentative data analysis (not shown in this work) has been performed to understand the nature of the reorientational motions at this temperature.51 Two diffusion-like processes have been identified (typical of melted phases): one with a self-diffusion coefficient in the same order of magnitude of [BH4] anion diffusion reported for molten LiBH4,52 and one with a diffusion coefficient one order of magnitude larger, not yet attributed. Since hydrogen decomposition also started, no further analysis or separation of various scattering contributions has been undertaken for this temperature, due to the uncertain nature of the amount and composition of the decomposition products.
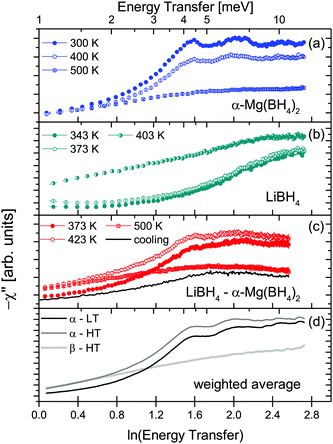 |
| | Fig. 15 −χ′′ as a function of ln(energy transfer) for (a) α-Mg(BH4)2, (b) LiBH4 and the mixture (c) LiBH4 + Mg(BH4)2. The lines in the bottom figure are the calculated weighted average, 2/3 Mg(BH4)2 + 1/3 LiBH4, of the pure compounds. The vertical error bars denote ±1σ. | |
5 Conclusions
The complex borohydrides Mg(BH4)2, LiBH4, and their 1:1 molar mixture have been investigated using neutron spectroscopy. For all investigated samples, the low energy transfer spectra are characterized by quasielastic features originating from jump rotation processes, and by vibrational lattice modes. The detailed dynamic response is closely linked to the respective crystal modification. Most striking is the difference between the dynamic properties of α- and β-Mg(BH4)2. Although the local Mg–[BH4] coordination in both phases is quite similar and jump rotations around the same symmetry axis are observed, the long range coordination plays a decisive role for the lattice dynamics and the activated rotation motions of the [BH4] tetrahedra. Vice versa, the entropic contribution of the [BH4] units and of the lattice dynamics at high temperatures might be at the origin for the metastability of the β-polymorph upon cooling. In LiBH4, the first order structural phase transition is accompanied by a change in the local reorientation of the [BH4] units, described as order–disorder transition. Notably, the [BH4] rotational symmetry already changes below the phase transition temperature, reminiscent of the pre-transition behaviour observed employing Raman spectroscopy.53 The ball milled mixture of LiBH4 + α-Mg(BH4)2 remains a physical mixture of the two compounds up to the melting temperature, and the characteristic dynamics of each constituent can be observed.
Conflicts of interest
There are no conflicts to declare.
References
- A. Züttel, P. Wenger, S. Rentsch, P. Sudan, P. Mauron and C. Emmenegger, J. Power Sources, 2003, 118, 1–7 CrossRef.
- S. Orimo, Y. Nakamori, G. Kitahara, K. Miwa, N. Ohba, S. Towata and A. Züttel, J. Alloys Compd., 2005, 404–406, 427–430 CrossRef CAS.
- J.-H. Her, P. W. Stephens, Y. Gao, G. L. Soloveichik, J. Rijssenbeek, M. Andrus and J.-C. Zhao, Acta Crystallogr., Sect. B: Struct. Sci., 2007, 63, 561–568 CrossRef CAS.
- K. Chłopek, C. Frommen, A. Léon, O. Zabara and M. Fichtner, J. Mater. Chem., 2007, 17, 3496–3503 RSC.
- J.-H. Kim, S.-A. Jin, J.-H. Shim and Y. W. Cho, Scr. Mater., 2008, 58, 481–483 CrossRef CAS.
- V. Ozolins, E. H. Majzoub and C. Wolverton, Phys. Rev. Lett., 2008, 100, 135501 CrossRef CAS PubMed.
- J. Voss, J. S. Hummelshøj, Z. Oodziana and T. Vegge, J. Phys.: Condens. Matter, 2009, 21, 012203 CrossRef CAS PubMed.
- X.-F. Zhou, A. R. Oganov, G.-R. Qian and Q. Zhu, Phys. Rev. Lett., 2012, 109, 245503 CrossRef PubMed.
- O. Zavorotynska, A. El-Kharbachi, S. Deledda and B. C. Hauback, Int. J. Hydrogen Energy, 2016, 41, 14387–14403 CrossRef CAS.
- Y. Filinchuk, B. Richter, T. R. Jensen, V. Dmitriev, D. Chernyshov and H. Hagemann, Angew. Chem., Int. Ed. Engl., 2011, 50, 11162–11166 CrossRef CAS PubMed.
- F. Buchter, Z. Oodziana, A. Remhof, O. Friedrichs, A. Borgschulte, P. Mauron, A. Züttel, D. Sheptyakov, G. Barkhordarian, R. Bormann, K. Chłopek, M. Fichtner, M. Sørby, M. Riktor, B. Hauback and S. Orimo, J. Phys. Chem. B, 2008, 112, 8042–8048 CrossRef CAS PubMed.
- Y. Filinchuk, E. Rönnebro and D. Chandra, Acta Mater., 2009, 57, 732–738 CrossRef CAS.
- T. Noritake, M. Aoki, M. Matsumoto, K. Miwa, S. Towata, H.-W. Li and S. Orimo, J. Alloys Compd., 2010, 491, 57–62 CrossRef CAS.
- I. Llamas-Jansa, O. Friedrichs, M. Fichtner, E. G. Bardaji, A. Züttel and B. C. Hauback, J. Phys. Chem. C, 2012, 116, 13472–13479 CrossRef CAS.
- Y. Filinchuk, D. Chernyshov and R. Černý, J. Phys. Chem. C, 2008, 112, 10579–10584 CrossRef CAS.
- P. Fischer and A. Züttel, Mater. Sci. Forum, 2004, 287–290 CAS.
- G. Renaudin, S. Gomes, H. Hagemann, L. Keller and K. Yvon, J. Alloys Compd., 2004, 375, 98–106 CrossRef CAS.
- Z. Oodziana and M. J. van Setten, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 81, 024117 CrossRef.
- N. Verdal, M. R. Hartman, T. Jenkins, D. J. DeVries, J. J. Rush and T. J. Udovic, J. Phys. Chem. C, 2010, 114, 10027–10033 CrossRef CAS.
- N. Verdal, T. J. Udovic and J. J. Rush, J. Phys. Chem. C, 2012, 116, 1614–1618 CrossRef CAS.
- T. J. Udovic, N. Verdal, J. J. Rush, D. J. De Vries, M. R. Hartman, J. J. Vajo, A. F. Gross and A. V. Skripov, J. Alloys Compd., 2013, 580, S47–S50 CrossRef CAS.
- N. Verdal, T. J. Udovic, W. Zhou, J. J. Rush, D. J. De Vries and M. R. Hartman, J. Phys. Chem. C, 2013, 117, 876–883 CrossRef CAS.
- A. Borgschulte, R. Gremaud, A. Züttel, P. Martelli, A. Remhof, A. J. Ramirez-Cuesta, K. Refson, E. G. Bardají, W. Lohstroh, M. Fichtner, H. Hagemann and M. Ernst, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 83, 024102 CrossRef.
- Y. Nakamori, K. Miwa, A. Ninomiya, H. Li, N. Ohba, S. Towata, A. Züttel and S. Orimo, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 74, 045126 CrossRef.
- D. Ravnsbaek, Y. Filinchuk, Y. Cerenius, H. Jakobsen, F. Besenbacher, J. Skibsted and T. Jensen, Angew. Chem., 2009, 121, 6787–6791 CrossRef.
- L. H. Rude, T. K. Nielsen, D. B. Ravnsbaek, U. Bösenberg, M. B. Ley, B. Richter, L. M. Arnbjerg, M. Dornheim, Y. Filinchuk, F. Besenbacher and T. R. Jensen, Phys. Status Solidi A, 2011, 208, 1754–1773 CrossRef CAS.
- E. Nickels, M. Jones, W. David, S. Johnson, R. Lowton, M. Sommariva and P. Edwards, Angew. Chem., Int. Ed. Engl., 2008, 47, 2817–2819 CrossRef CAS PubMed.
- P. Schouwink, V. D'Anna, M. B. Ley, L. M. Lawson Daku, B. Richter, T. R. Jensen, H. Hagemann and R. Černý, J. Phys. Chem. C, 2012, 116, 10829–10840 CrossRef CAS.
- G. Capurso, F. Agresti, L. Crociani, G. Rossetto, B. Schiavo, A. Maddalena, S. L. Russo and G. Principi, Int. J. Hydrogen Energy, 2012, 37, 10768–10773 CrossRef CAS.
- E. G. Bardají, Z. Zhao-Karger, N. Boucharat, A. Nale, M. J. van Setten, W. Lohstroh, E. Röhm, M. Catti and M. Fichtner, J. Phys. Chem. C, 2011, 115, 6095–6101 CrossRef.
- Z.-Z. Fang, X.-D. Kang, P. Wang, H.-W. Li and S.-I. Orimo, J. Alloys Compd., 2010, 491, L1–L4 CrossRef CAS.
- L. Zhang, J. Zheng, X. Xiao, X. Fan, X. Huang, X. Yang and L. Chen, RSC Adv., 2017, 7, 36852–36859 RSC.
- N. Verdal, T. J. Udovic and J. J. Rush, J. Phys. Chem. C, 2012, 116, 1614–1618 CrossRef CAS.
- N. Verdal, T. J. Udovic, J. J. Rush, H. Wu and A. V. Skripov, J. Phys. Chem. C, 2013, 117, 12010–12018 CrossRef CAS.
- A. Remhof, Z. Oodziana, P. Martelli, O. Friedrichs, A. Züttel, A. V. Skripov, J. P. Embs and T. Strässle, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 81, 214304 CrossRef.
- R. Gremaud, A. Züttel, A. Borgschulte, A. Ramirez-Cuesta, K. Refson and D. Colognesi, Chem. Phys., 2013, 427, 22–29 CrossRef CAS.
- J.-P. Soulié, G. Renaudin, R. Černý and K. Yvon, J. Alloys Compd., 2002, 346, 200–205 CrossRef.
- F. Buchter, Z. Oodziana, P. Mauron, A. Remhof, O. Friedrichs, A. Borgschulte, A. Züttel, D. Sheptyakov, T. Strässle and A. J. Ramirez-Cuesta, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 78, 094302 CrossRef.
- Y. Filinchuk, R. Černý and H. Hagemann, Chem. Mater., 2009, 21, 925–933 CrossRef CAS.
- A. V. Skripov, A. V. Soloninin, O. A. Babanova, H. Hagemann and Y. Filinchuk, J. Phys. Chem. C, 2010, 114, 12370–12374 CrossRef CAS.
- A. V. Soloninin, O. A. Babanova, A. V. Skripov, H. Hagemann, B. Richter, T. R. Jensen and Y. Filinchuk, J. Phys. Chem. C, 2012, 116, 4913–4920 CrossRef CAS.
- D. T. Shane, L. H. Rayhel, Z. Huang, J.-C. Zhao, X. Tang, V. Stavila and M. S. Conradi, J. Phys. Chem. C, 2011, 115, 3172–3177 CrossRef CAS.
- W. Lohstroh and Z. Evenson, Journal of large-scale research facilities, 2015, 1, 1–3 CrossRef.
- T. Unruh, J. Neuhaus and W. Petry, Nucl. Instrum. Methods Phys. Res., Sect. A, 2007, 580, 1414–1422 CrossRef CAS.
- L. Silvi, E. Röhm, M. Fichtner, W. Petry and W. Lohstroh, Phys. Chem. Chem. Phys., 2016, 18, 14323–14332 RSC.
-
R. Hempelmann, Quasielastic Neutron Scattering and Solid State Diffusion, Clarendon Press, 2000 Search PubMed.
- K. Sköld, J. Chem. Phys., 1968, 49, 2443–2445 CrossRef.
- A. Giannasi, D. Colognesi, L. Ulivi, M. Zoppi, A. J. Ramirez-Cuesta, E. G. Bardají, E. Röhm and M. Fichtner, J. Phys. Chem. A, 2010, 114, 2788–2793 CrossRef CAS PubMed.
- H. Hagemann, V. D'Anna, J.-P. Rapin, R. Černý, Y. Filinchuk, K. C. Kim, D. S. Sholl and S. F. Parker, J. Alloys Compd., 2011, 509(Suppl. 2), S688–S690 CrossRef CAS.
- Y. Filinchuk, R. Černý and H. Hagemann, Chem. Mater., 2009, 21, 925–933 CrossRef CAS.
-
L. Silvi, PhD thesis, Technische Universität München, 2017.
- P. Martelli, A. Remhof, A. Borgschulte, P. Mauron, D. Wallacher, E. Kemner, M. Russina, F. Pendolino and A. Züttel, J. Phys. Chem. A, 2010, 114, 10117–10121 CrossRef CAS PubMed.
- H. Hagemann, Y. Filinchuk, D. Chernyshov and W. van Beek, Phase Transitions, 2009, 82, 344–355 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8cp04316g |
| ‡ Present address: Institut für Weiche Materie und funktionale Materialien (EMISFM), Helmholtz-Zentrum Berlin für Materialien und Energie, Hahn Meitner Platz 1, 14109 Berlin, Germany. |
|
| This journal is © the Owner Societies 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *a,
Zhirong
Zhao-Karger
*a,
Zhirong
Zhao-Karger
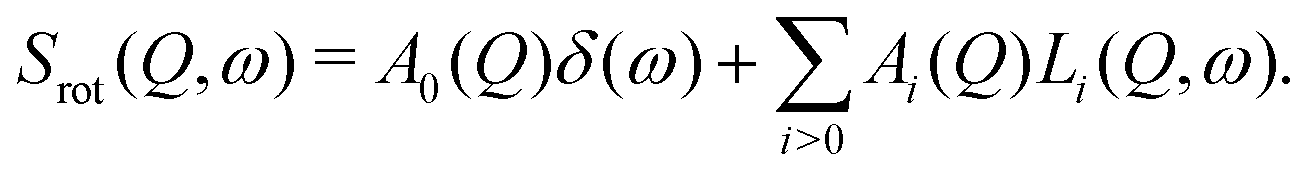
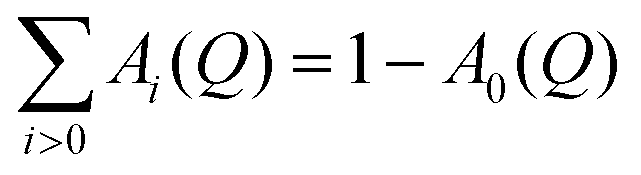 . A0(Q) is the elastic incoherent structure factor of the rotation which depends on the spatial extension and its symmetry. For simple rotations, expressions for A0(Q) can be obtained analytically, see for example ref. 46.
. A0(Q) is the elastic incoherent structure factor of the rotation which depends on the spatial extension and its symmetry. For simple rotations, expressions for A0(Q) can be obtained analytically, see for example ref. 46.
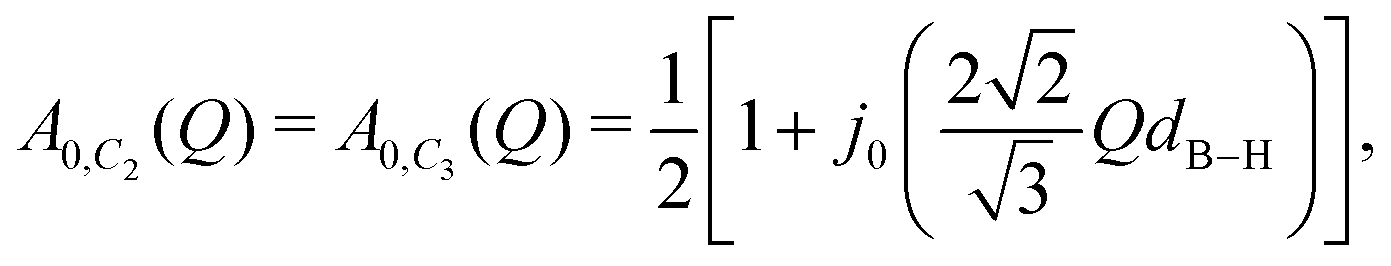
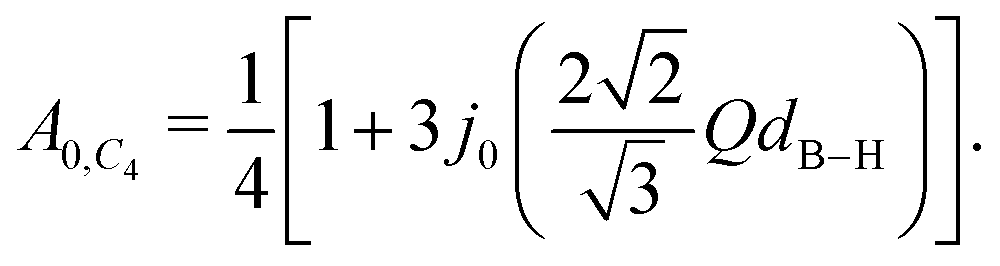
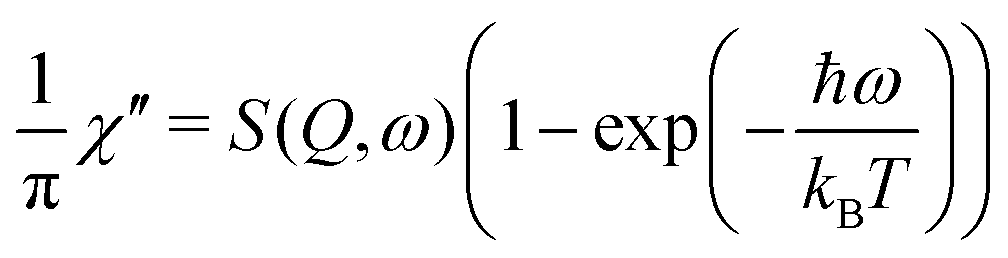 , with kB the Boltzmann constant. Data were summed over Q in the 1.5 ≤ Q ≤ 4.25 Å−1 range. For α-Mg(BH4)2, distinct peaks at around 4.2 and 8 meV are visible which hardly change with temperature. In contrast, the β-polymorph has slightly softer low energy excitations and the features are much broader and less defined. The plot underlines that β-Mg(BH4)2 shows more dynamical disorder compared to the α-phase, starting already at temperatures as low as 200 K. This is also reflected in the mean square displacement 〈u2〉, as illustrated in Fig. 9. The mean square displacement of α-Mg(BH4)2 follows a linear relationship with increasing temperature. In the β-modification, 〈u2〉 follows approximately the same relationship below 200 K, but between 200 and 300 K, a jump like increase is observed. In the same temperature region, the jump rotation processes in β-Mg(BH4)2 become fully activated45 whereas the rotation modes in α-Mg(BH4)2 are still significantly hindered even at 400 K. Incidentally, the anomalous lattice expansion observed in β-Mg(BH4)2 shows a change from contraction to the expansion of the a-lattice parameter around 200 K and the opposite for the b-axis lattice parameter, indicating a link between the local rotational dynamics, lattice dynamics and structure. In α-Mg(BH4)2, anomalous expansion is much smaller and it is restricted to temperatures below 200 K.39
, with kB the Boltzmann constant. Data were summed over Q in the 1.5 ≤ Q ≤ 4.25 Å−1 range. For α-Mg(BH4)2, distinct peaks at around 4.2 and 8 meV are visible which hardly change with temperature. In contrast, the β-polymorph has slightly softer low energy excitations and the features are much broader and less defined. The plot underlines that β-Mg(BH4)2 shows more dynamical disorder compared to the α-phase, starting already at temperatures as low as 200 K. This is also reflected in the mean square displacement 〈u2〉, as illustrated in Fig. 9. The mean square displacement of α-Mg(BH4)2 follows a linear relationship with increasing temperature. In the β-modification, 〈u2〉 follows approximately the same relationship below 200 K, but between 200 and 300 K, a jump like increase is observed. In the same temperature region, the jump rotation processes in β-Mg(BH4)2 become fully activated45 whereas the rotation modes in α-Mg(BH4)2 are still significantly hindered even at 400 K. Incidentally, the anomalous lattice expansion observed in β-Mg(BH4)2 shows a change from contraction to the expansion of the a-lattice parameter around 200 K and the opposite for the b-axis lattice parameter, indicating a link between the local rotational dynamics, lattice dynamics and structure. In α-Mg(BH4)2, anomalous expansion is much smaller and it is restricted to temperatures below 200 K.39