
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Magnetic structure of UO2 and NpO2 by first-principle methods†‡
James T.
Pegg
 *ab,
Ashley E.
Shields
c,
Mark T.
Storr
b,
Andrew S.
Wills
a,
David O.
Scanlon
ade and
Nora H.
de Leeuw
af
*ab,
Ashley E.
Shields
c,
Mark T.
Storr
b,
Andrew S.
Wills
a,
David O.
Scanlon
ade and
Nora H.
de Leeuw
af
aDepartment of Chemistry, University College London, 20 Gordon Street, London WC1H 0AJ, UK. E-mail: uccajtp@ucl.ac.uk
bAtomic Weapons Establishment (AWE) Plc, Aldermaston, Reading, RG7 4PR, UK
cOak Ridge National Laboratory, One Bethel Valley Road, Oak Ridge, Tennessee 37831, USA
dDiamond Light Source Ltd., Diamond House, Harwell Science and Innovation Campus, Didcot, Oxfordshire OX11 0DE, UK
eThomas Young Centre, University College London, Gower Street, London WC1E 6BT, UK
fCardiff University, School of Chemistry, Main Building, Park Place, Cardiff, CF1D 3AT, UK
First published on 10th December 2018
Abstract
The magnetic structure of the actinide dioxides (AnO2) remains a field of intense research. A low-temperature experimental investigation of the magnetic ground-state is complicated by thermal energy released from the radioactive decay of the actinide nuclei. To establish the magnetic ground-state, we have employed high-accuracy computational methods to systematically probe different magnetic structures. A transverse 1k antiferromagnetic ground-state with Fmmm (No. 69) crystal symmetry has been established for UO2, whereas a ferromagnetic (111) ground-state with R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m (No. 166) has been established for NpO2. Band structure calculations have been performed to analyse these results.
m (No. 166) has been established for NpO2. Band structure calculations have been performed to analyse these results.
1. Introduction
The magnetic ground-state of the actinide dioxides (AnO2) is key to the design of reliable computational models.1 The major actinides (An = U, Np, Pu) are challenging systems to study. The low-temperature characterisation of the AnO2 magnetic ground-state is complicated by: the toxicity of the metals,2–4 nucleonic radioactive decay,2–4 and the inhomogeneity of samples.5–10 To compensate for known radiogenic and experimental issues, computational methods offer a complementary means of investigation.A number of experimental studies on the AnO2 indicate Fmm (No. 225) cubic symmetry,11,12 where the An4+ cations occupy octahedral (4a) sites and the O2− anions occupy tetrahedral (8c) sites. Low-temperature measurements of UO2 have indicated the Pa (No. 205) crystal symmetry, which involves an internal distortion of the O2− ions within the cubic lattice.13 The magnetic structure of the AnO2 is often inferred by the one-electron crystal electric field (Fig. 1). In crystal-field (CF) theory, the 5f electrons are highly localized due to the insulating nature of these materials. Thus, the orbitals do not hybridize and the crystal field is influenced by the electrostatic potential. Therefore low-spin coupling is initially considered, with the crystal field as a perturbation. The spin–orbit interaction (SOI)14 generates j = 7/2 and j = 5/2 electronic levels, whereby the degeneracy of the levels is further broken by the crystal field. The interpretation of the magnetic structure by CF theory is only valid for the one-electron case,15,16 whereas the magnetic structure of the AnO2 involves the complex interplay of spin–lattice, magneto-elastic, super-exchange, multipolar and cooperative Jahn–Teller interactions.17–21 The type of magnetism can be classified into paramagnetic (PM); diamagnetic (DM); ferromagnetic (FM); anti-ferromagnetic (AFM); and ferrimagnetic (FI) behaviour.1
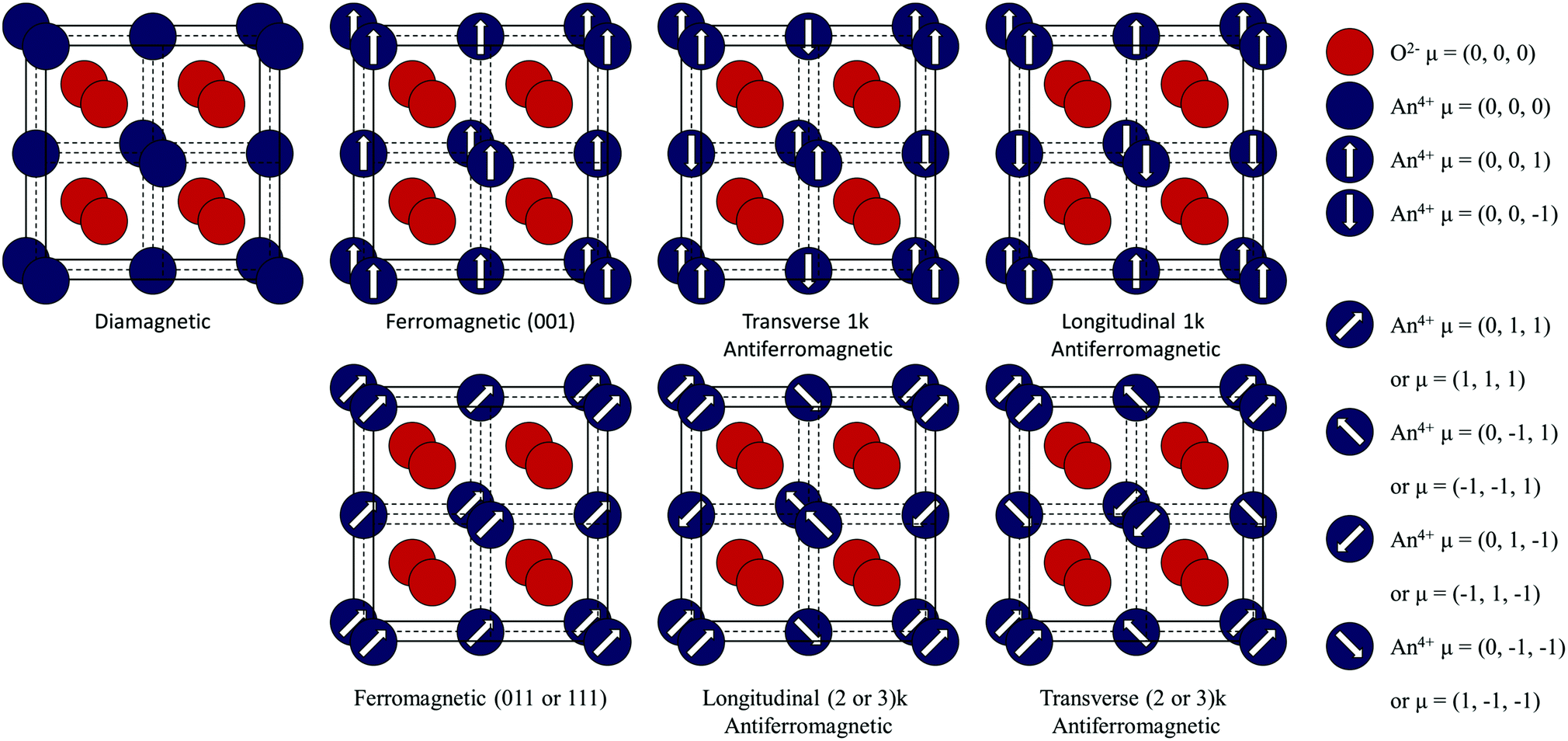 | ||
| Fig. 1 Illustration of magnetic configurations in the calcium fluorite (CaF2) crystal structure. | ||
In DM materials, as all electrons are paired, no magnetic moments are associated with the individual ions and as a result the net magnetic moment of the crystal is zero. The only magnetic response is a weak repulsion in an applied magnetic field. In ordered FM, AFM or FI materials, a magnetic moment is formed by unpaired electrons. These magnetic moments may couple resulting in a periodic arrangement. If the magnetic moments are aligned in one dimension, the material is FM and has a net crystal moment. The direction of the magnetic moment may vary, resulting in FM (111), (011) or (001) states in the Fmm crystal. If the magnetic moments are opposed, the material is AFM and no net magnetic moment exists. In the propagation vector formulism, contributions can be combined from a number of symmetry-related wavevectors. These are termed multi-k structures.1 In the following article, single (1k), double (2k), and triple (3k) structures have been considered.
If the opposing magnetic moments on the ions are unequal, the material is FI and the crystal has a net magnetic moment. If the magnetic moments are decoupled, the material is PM and there is no ordered distribution. The net magnetic moment of the crystal will average to zero. In addition, an isolated ion is said to be PM if it has a magnetic moment.22 As the magnetic moment of the An4+ ions (in stoichiometric AnO2) are equal (due to the occupation of chemically equivalent sites) the FI state cannot exist. In addition, significant exchange interactions are expected to be present and cause magnetic order. As a result, neither FI or PM is considered in this study. The manifestation of metastable states and the juxtaposition of energy levels makes the determination of the magnetic ground-state challenging. In this paper, the magnetic structures of the AnO2 are calculated for multiple configurations (Table 1).
| Ion | Ferromagnetic | Antiferromagnetic (longitudinal domain) | Antiferromagnetic (transverse domain) | ||||||
|---|---|---|---|---|---|---|---|---|---|
| (001) | (011) | (111) | 1k | 2k | 3k | 1k | 2k | 3k | |
| (0, 0, 0) | (0, 0, 1) | (0, 1, 1) | (1, 1, 1) | (0, 0, 1) | (0, 1, 1) | (1, 1, 1) | (0, 0, 1) | (0, 1, 1) | (1, 1, 1) |
| (½, ½, 0) | (0, 0, 1) | (0, 1, 1) | (1, 1, 1) | (0, 0, 1) | (0, ![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , 1) , 1) |
(, , 1) |
(0, 0, ) |
(0, , ) |
(1, , ) |
| (½, 0, ½) | (0, 0, 1) | (0, 1, 1) | (1, 1, 1) | (0, 0, ) |
(0, 1, ) |
(, 1, ) |
(0, 0, 1) | (0, , 1) |
(, , 1) |
| (0, ½, ½) | (0, 0, 1) | (0, 1, 1) | (1, 1, 1) | (0, 0, ) |
(0, , ) |
(1, , ) |
(0, 0, ) |
(0, 1, ) |
(, 1, ) |
The magnetic wavevectors are directed along the main axes of the crystal unit cell. The final magnetic moment of each ion is calculated using the Pythagorean theorem in eqn (1).
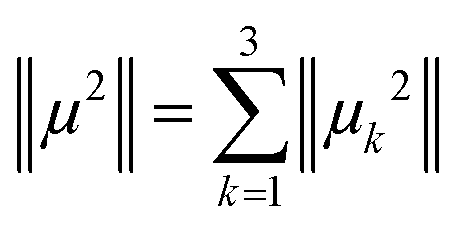 | (1) |
The crystal structure is coupled by SOI14 to the magnetic state. In the high-temperature PM state, the Fmm (No. 255) crystal structure is stabilized by the intrinsic magnetic disorder (Fig. 2). At low-temperature (T = 30.8 K), the ordered FM and AFM configurations can cause a crystallographic distortion due to an imbalance of magnetic forces. The extent of the disruption is dependent upon the magnitude of the magnetic forces within the crystal. Low-temperature (T < 30.8 K) measurements of the AnO2 magnetic ground-state are extremely challenging, due to: thermal energy generated by the radioactive decay of the actinide nuclei, the inhomogeneity of samples and high chemical sensitivity to environmental conditions.23–30 In this paper, the low-temperature magnetic structure of UO2 and NpO2 is investigated using first-principle methods. In addition, a computationally tractable method for the inclusion of magnetic order in the AnO2 has been developed that can be applied to larger systems.132
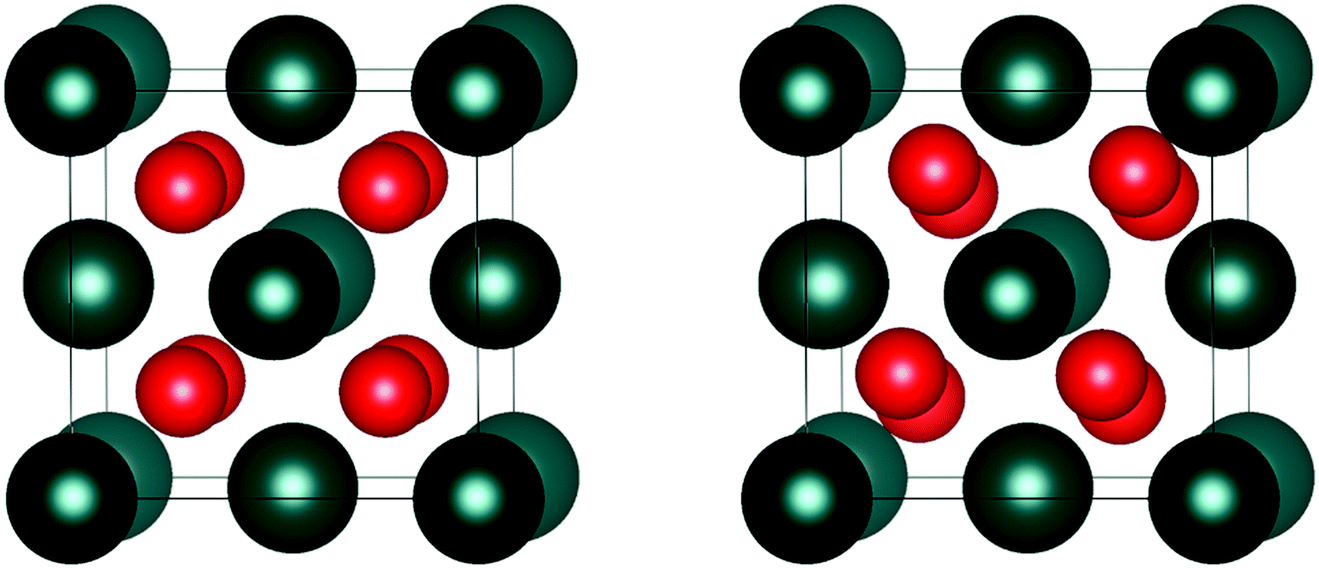 | ||
| Fig. 2 Illustration of the actinide dioxide (AnO2) crystal structure: (left) cubic Fmm symmetry, (right) cubic Pa symmetry. The colours in the parentheses indicate the An4+ (blue) and O2− (red) ions. | ||
A key difficulty in the computational investigation of the AnO2 is the treatment of: relativistic influences, electron-correlation, and complex magnetic structures. The correct electronic structure of the AnO2 cannot be calculated by conventional density functional theory (DFT) based methods,31,32 due to the high degree of electron-correlation,33 which often manifests as an underestimation of the electronic band-gap.9,34 To compensate, numerous methods have been developed such as, the self-interaction correction (SIC) method,35 modified density functional theory (DFT+U),31,32,36–38 dynamic mean field theory (DMFT),39 and hybrid density functionals.40–42 Of these methods, DFT+U offers a computationally tractable means of investigation by treating the on-site Coulomb repulsion of the f-electrons with tuneable U and J modifiers, essentially adding an energy penalty to partial occupation of what should be localized f-electrons. The ground-state characteristics of f-electron compounds can be captured by DFT+U when SOI are included.43 By comparison, hybrid functionals offer one of the more accurate, but computationally highly expensive methods. The computationally intense nature of DMFT has been highlighted by a number of authors, but even this method has incorrectly calculated the early AnO2 as charge-transfer insulators.39,44–47 In addition to the highly-correlated and relativistic nature of actinide compounds, the identification and modelling of the magnetic ground-state is incredibly challenging. A limited number of publications have investigated noncollinear contributions,48–50 and collinear 1k AFM order is often used to model the electronic structure.9,34,42,49,51–63 The inclusion of spin–orbit interactions (SOI)14 is often ignored,34,46,48,60–62,64 although a number of investigations have highlighted its importance.33,48 This is mainly due to the computational cost.63
The magnetic structure of UO2 displays interesting characteristics. In the Russell–Saunders (low-spin) coupling scheme, the ground-state of the U4+ ion is a 3H4 (Γ5 triplet) multiplet.15,16,48 Low-temperature measurements of (U1−xThx)O2 confirm a Γ5 triplet magnetic ground-state.65 A discontinuous first-order phase transition (Néel temperature, TN = 30.8 K66) that is indicative of magnetic order has been confirmed by: heat capacity,67,68 magnetic susceptibility5 and neutron diffraction69–71 measurements. The nature of the magnetic ground-state has been investigated by numerous authors. Initial neutron diffraction measurements indicated a collinear 1k AFM15,16 ground-state, coupled with a homogeneous lattice distortion.19 Later studies suggested an internal Jahn–Teller distortion66,71–74 of the O2− ions, but no evidence has been found for a reduction in the external cubic crystal symmetry.69,71,74,75 On further investigation, noncollinear 2k AFM order was proposed due to internal crystallographic distortion.71,74
Finally, low-temperature (T < 30.8 K) neutron diffraction measurements of UO2 have confirmed an internal Pa (No. 205) crystallographic distortion, where the displacement of the O2− ions is 0.014 Å,13,20,71,74 which is indicative of transverse 3k AFM order.13,33,76 Anti-ferroquadrupolar ordering favours Pa (No. 205) crystal symmetry by minimizing quadrupolar and exchange terms.18 Neutron diffraction measurements have determined an ordered effective magnetic moment of 1.74 μB per U ion,71 whereas the transverse 3k AFM ground-state has been inferred by inelastic neutron scattering (INS),77 resonant X-ray scattering (RXS)72 and nuclear magnetic resonance (NMR)20 measurements. In terms of the electronic structure, the f–f Mott–Hubbard insulating character of UO2 with a band-gap of 2.00–2.50 eV2,78,79 has been established by optical adsorption,2,79 X-ray adsorption (XAS),3,78,80 X-ray photoemission (XPS),81–84 bremsstrahlung isochromatic spectroscopy (BIS),83,84 resonant photoemission spectroscopy (RPES),85 inverse photoemission spectroscopy (IPES)86 and theoretical methods.48,87,88 In the past, multiple investigations of UO2 have focused on the Fmm (No. 225) crystal structure,11,18,69,80,89,90 but in this study, in addition to the experimental AFM ground-state, the energetics of the DM and FM configurations have been considered for comparison with previous theoretical considerations.91,92
By comparison, the magnetic ground-state of NpO2 is highly complex and marred by several inconsistencies.22 In crystal field theory, the Np4+ ion results in the 4I9/2 (Γ8 quartet) configuration. Indeed, the tetravalent f3 nature of the Np4+ ion has been confirmed by Mossbauer isomer shift spectroscopy.93 At low-temperature (T = 25.4 K), a first-order PM-AFM phase transition has been indicated by magnetic susceptibility94 and specific heat capacity measurements.95,96 In addition, a longitudinal 3k AFM ground-state coupled to Fmm (No. 225) crystal symmetry has been inferred from resonant X-ray scattering73 (10 K < T < 17 K) and 17O NMR measurements (T = 17 K).97 The absence of an external distortion of the cubic cell further indicates noncollinear 3k AFM behavior.96 In contrast, low-temperature Mossbauer (T = 1.5 K)93 and neutron diffraction (12 K < T < 30 K)98 measurements have failed to identify a magnetic moment. The upper limit for the magnetic moment set by muon spin rotation measurements (0.3 K < T < 25.4 K) is 0.06–0.15 μB per Np ion,22,99 whereas the upper limit for the magnetic moment set by Mössbauer spectroscopy is 0.01 μB per Np ion.93 The absence of a measurable local magnetic moment is inconsistent with the idea of magnetic order and the nature of the small-moment AFM state is unresolved.22 As a Kramers ion, i.e. an ion with an uneven number of valence electrons, the ground Np4+ state should order magnetically in the absence of interactions that break time-reversal symmetry conditions. A mechanism by which the magnetic moment of the Np4+ ion can be suppressed is AFM super-exchange,22 which is the coupling between moment-bearing cations via nominally DM anions.100,101
The inhomogeneous nature of NpO2 samples has hindered experimental investigation.96,102 For instance, the extreme difficulty in manufacturing large single-crystal samples impacts the search for low-temperature (T < 25.4 K) crystallographic distortions.96,103 In the past, the detection threshold of neutron diffraction measurements was limited to 0.02–0.03 Å.22 By comparison, crystallographic distortions of isostructural UO2 are of the order of 0.01–0.02 Å.19 In a search for low-temperature (T < 25.4 K) anharmonic effects in NpO2 by neutron diffraction (12 K < T < 30 K)98 and in an independent RXS (9 K < T < 25 K) study,96 no evidence of a dynamical distortion of the O2− ions was found. However, Mossbauer spectroscopy (T = 1.5 K) of NpO2 has indicated an internal O2− ion distortion inferred by the small broadening of spectroscopic lines.93 In addition, inelastic neutron scattering (INS) (5 K < T < 25 K) studies104,105 indicate an internal O2− ion distortion of 0.02 Å, which is reminiscent of the internal O2− ion distortion in UO2. The internal O2− ion distortion may result in Pa (No. 205) crystal symmetry that is indicative of transverse 3k AFM behaviour, although this has yet to be experimentally confirmed.
The upper-limit of the magnetic moment (0.01–0.15 μB per Np ion) indicates that NpO2 is a small-moment system.22,93,96,99 To our knowledge, small moments have only been identified in heavy-fermion metals106–108 and has yet to be identified in insulators. In this regard, computational investigations of PuO2 have indicated the existence of an unconfirmed small-moment insulating magnetic ground-state.1 For instance, multiple DFT calculations have indicated an AFM ground-state, which contrasts with the DM state established by experimental methods.109–112 The mechanisms behind such an intriguing electronic states are not yet fully understood; where, the crystal and magnetic structures of NpO2 remain unresolved.
2. Computational methodology
A noncollinear relativistic computational study of the AnO2 (An = U, Np) magnetic structure by the Vienna Ab initio Simulation Package (VASP) code has been conducted.35,39,113 The investigation considers: hybrid Heyd–Scuseria–Ernzerhof (HSE06),40–42 PBEsol+U,36–38 and PBEsol31,32 functionals.31,32,36–38 A planewave basis set with a kinetic energy cut-off of 500 eV has been used. The following valence electrons have been considered for: uranium (6s2, 7s2, 6p6, 6d2 5f2), neptunium (6s2, 7s2, 6p6, 6d2 5f3), and oxygen (2s2, 2p4). The influence of noncollinear magnetic behaviour and SOI14 has also been included. The spin quantisation axis is defined by (0, 0, 1) plane, from which magnetic and spinor-like values are calculated. An ionic relaxation with the conjugate gradient algorithm has been completed.114 The space group has been evaluated to 10−5 Å based on a symmetry analysis of the unit cell. Images are visualized by the VESTA code.115As a computationally intensive method, hybrid functionals incorporate Hartree–Fock (HF) exchange energy into the DFT formulism. In this study, the hybrid HSE06 functional has been used.40–42,116 The integration of the Brillouin zone has been calculated from a Γ-centred (4·4·4) k-point grid with the conventional Gaussian method.117
| EHSEXC = (a)EHF,SRX(μ) + (1 − a)EPBE,SRX(μ) + EPBE,LRX(μ) + EPBEC | (2) |
To capture the highly-correlated nature of f-electron compounds, DFT+U offers a more computationally tractable method.31,32,36–38 The on-site Coulomb repulsion of the An 5f-electrons has been treated by the rotationally invariant Liechtenstein et al. DFT+U31,32,36–38 formulism,37 where the Coulomb (U) and exchange (J) modifiers are treated as independent variables:
 | (3) |
In an earlier study on the AnO2, the performance of multiple DFT functionals was tested;33 where, the PBEsol functional had been proven to be the most effective. The exchange–correlation energy is therefore evaluated by the PBEsol functional.119 The integration of the Brillouin zone is performed with a Γ-centred (5·5·5) k-point grid,117 using the Blöchl tetrahedron method.120 Self-consistent calculations were performed until convergence was reached for the respective electronic and ionic thresholds of 1 × 10−8 eV and 1 × 10−3 eV A−1. For the optical absorbance calculation, the k-point mesh is 15·15·15; for band structure calculations, the Fmmm (No. 69) k-point pathway is Γ → Y → X → Z → Γ → L, whereas, the Pa (No. 205) k-point pathway is Γ → M → R → X → Γ → R.
3. Results & discussions
3.1 Uranium dioxide
(No. 205) distortion of the transverse 3k AFM state agrees with neutron diffraction measurements.13,20,71,74
| Initial configuration | Relative energy (eV) | Band-gap (eV) | Magnetic moment (μB per U ion) | Lattice volume (Å3) | Space group (number) | |
|---|---|---|---|---|---|---|
| Note: the initial DM HSE06 state is unstable; a relaxed, highly-energetic FM (852) state has been identified. | ||||||
| Diamagnetic | ||||||
| 0.659 | 1.75 | 0.85 | 161.70 |
Fmm (No. 225) |
||
| Ferromagnetic | ||||||
| (001) | 0.010 | 2.42 | 1.53 | 162.78 | I4/mmm (No. 139) | |
| (011) | 0.026 | 2.62 | 1.45 | 162.81 | Immm (No. 71) | |
| (111) | 0.012 | 2.57 | 1.44 | 162.60 |
Rm (No. 166) |
|
| Antiferromagnetic | ||||||
| Longitudinal | 1k | 0.006 | 2.79 | 1.51 | 162.63 | I4/mmm (No. 139) |
| 2k | 0.019 | 2.88 | 1.43 | 162.48 | I4/mmm (No. 139) | |
| 3k | 0.023 | 2.82 | 1.42 | 162.47 |
Fmm (No. 225) |
|
| Transverse | 1k | 0.000 | 3.02 | 1.49 | 162.53 | Fmmm (No. 69) |
| 2k | 0.000 | 3.02 | 1.49 | 162.55 | Pbca (No. 61) | |
| 3k | 0.014 | 2.99 | 1.43 | 162.57 |
Pa (No. 205) |
|
| Experimental | ||||||
| — | 2.00–2.5079,121 | 1.7469,71,74 | 163.8569,74 |
Pa (No. 205)13 |
||
To reduce the computational cost and to further probe the magnetic ground-state, the relative energetics of competing magnetic phases have been calculated with PBEsol+U. The relative energetics of the magnetic states are influenced by the U modifier (Fig. 3). In contrast to hybrid HSEE06 calculations, the highly-energetic PBEsol+U DM state is stable. In our calculations, the metallic FM (111) ground-state, calculated by PBEsol (U = 0 eV), is inconsistent with the experimental data. Indeed, under no circumstance is the insulating nature of UO2 reproduced when U = 0 eV, which illustrates the failure of pure DFT to account for the highly localized character of the 5f electrons. The insulating nature of UO2, which is well-described experimentally, can be reproduced computationally when U = 3–4 eV. The introduction of the U modifier immediately results in an AFM ground-state. The nature of the AFM ground-state is, however, dependent on the U constraint. A degenerate longitudinal 2–3k AFM and transverse 1–3k AFM ground-state is formed when U = 1–7 eV. In comparison with HSE06 calculations, the relative degeneracy of the transverse 1–2k AFM PBEsol+U ground-states has also been shown. In relation to past theory, the DM is considerably higher in energy and therefore physically unrealistic.
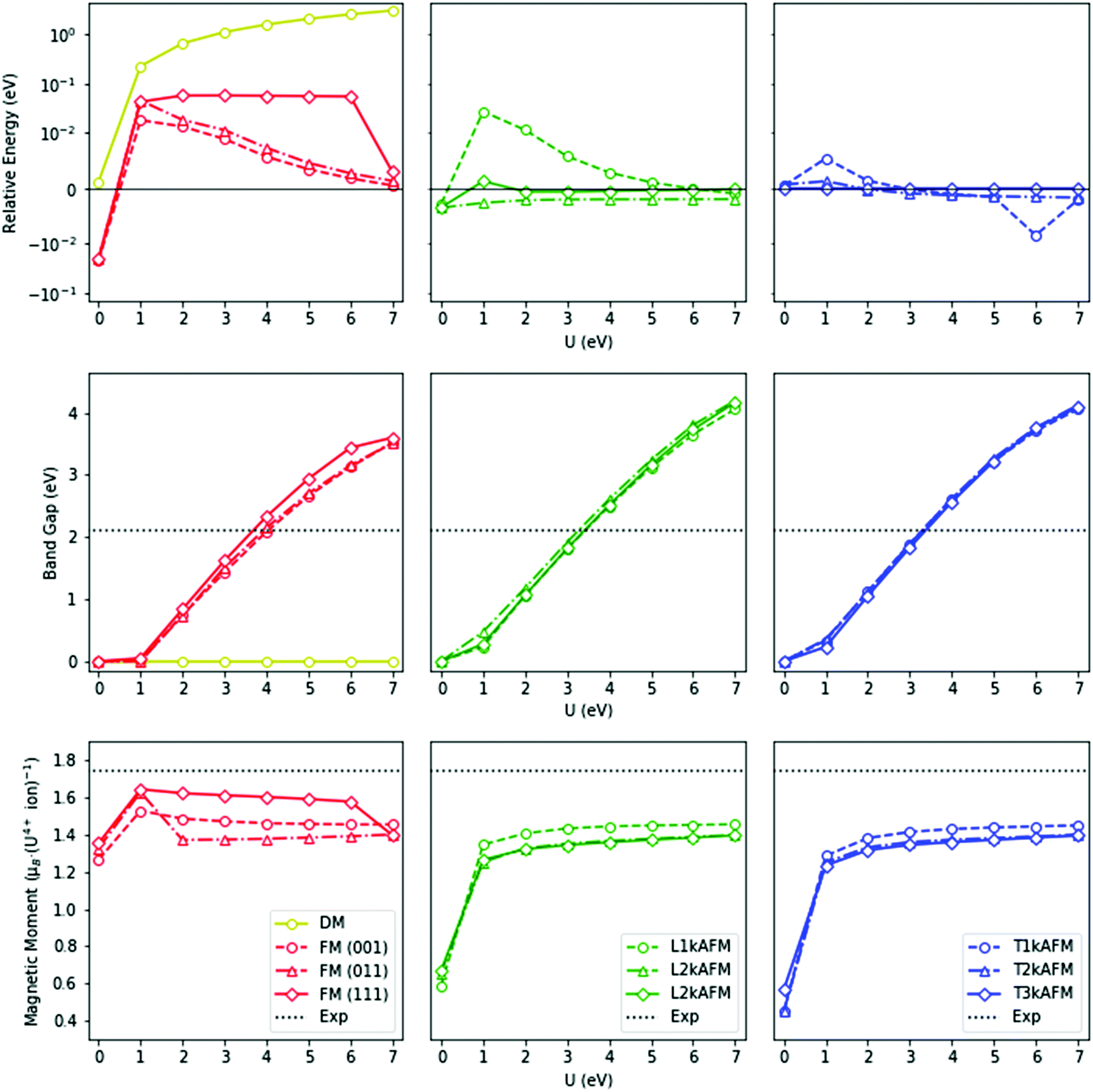 | ||
| Fig. 3 The relative ground-state energies, band-gaps, and effective magnetic moments against the Coulomb modifier (U) for diamagnetic (DM), ferromagnetic (FM), and antiferromagnetic (AFM) states of UO2, calculated with PBEsol+U. The antiferromagnetic transverse (T) and longitudinal (L) domains are also represented. The k-prefix denotes the number of independent wave vectors: above, the calculated energy of magnetic states relative to the transverse 3k antiferromagnetic state; middle, the fundamental band-gap; below, the effective magnetic moment of the uranium ions. The DM (yellow), FM (orange), longitudinal AFM (green) and transverse AFM (blue) states are indicated. | ||
The low-temperature crystal structure of UO2 shows Pa crystal symmetry indicated by neutron scattering and X-ray diffraction measurements.13 In our calculations, the transverse 3k AFM state results in cubic Pa (No. 205) crystal symmetry, and is consistent with experimental information.13 As a model of the cubic crystal structure at low-temperature, the collinear 1k AFM states are an invalid approximation. The transverse 1k AFM with orthorhombic Fmmm (No. 69) symmetry differs from the longitudinal 1k AFM state with tetragonal I4/mmm (No. 139) symmetry. The magnetic moment of UO2 is 1.74 μB per U ion, reported by neutron diffraction measurements.69,71,74 In our calculations, the magnetic moment of the U ion is underestimated by DFT+U and the hybrid HSE06 functional with the closest approximation obtained by the FM (111) states. In terms of the lattice volume, no discernible change is detected between the ordered magnetic states, but the lattice volume is considerably lower in the DM state.
 | ||
| Fig. 4 The electronic structure of UO2 calculated by PBEsol for the: (left) transverse 1k AFM state (U = 3.50 eV, J = 0.00 eV); (right) transverse 3k AFM state (U = 3.35 eV, J = 0.00 eV). The valence band (dark blue), conduction band (orange), U f- (blue), U d- (green) and O p- (red) states are indicated. | ||
In the density of states (DoS), the valence band maximum (VBM) and conduction band minimum (CBM) are mainly comprised of uranium f-states. The U d-states are significantly higher in energy and should therefore have very little influence on bonding interactions. This electronic structure indicates that UO2 is a Mott–Hubbard insulator, consistent with experimental information. The band structure of transverse 1k AFM UO2 ground-state results in a Γ (VBM) to Γ–Y (CBM) indirect fundamental band-gap of 2.27 eV; whereas the calculated optical absorption spectra band-gap is 2.49 eV. The fundamental band-gap and optical band-gap (although they are not strictly directly comparable) differ by 0.22 eV. The fundamental band-gap defines the VBM–CBM energy difference, whereas, the optical band-gap defines the minimum allowed transition as controlled by symmetry rules. The calculated band-gap and bulk modulus are in very good agreement with experimental information (Table 3). The electronic structure of the transverse 3k AFM is also shown; here, the degeneracy of the bands is noticeably perturbed by noncollinear order.
| DFT+U | Band-gap (eV) | Lattice constant (Å) | Bulk modulus (GPa) | Magnetic moment (μB per U ion) | Crystal symmetry | Magnetic state | ||
|---|---|---|---|---|---|---|---|---|
| U (eV) | J (eV) | Fundamental | Optical | |||||
| 3.50 | 0.00 | 2.27 | 2.49 | 5.476 | 210 | 1.42 | Fmmm (69) | T-1k AFM |
| 3.35 | 0.00 | 2.06 | 2.20 | 5.474 | 210 | 1.35 |
Pa (205) |
T-3k AFM33 |
| — | — | 2.00–2.5079,121 | ∼5.47369,74 | 20711 | 1.7469,71,74 |
Pa (205) |
Experimental | |
3.2 Neptunium dioxide
(No. 205) crystal symmetry, which satisfies observations of noncollinear magnetic behaviour and the inferred internal crystallographic distortion. However, the magnetic moment is anomalously high and does not fit the picture of a small-moment AFM state. As with UO2 and PuO2,1 the crystal symmetry and magnetic structure of NpO2 are coupled by SOI. The low temperature crystal structure of NpO2 is unresolved, but thus far, Pa (No. 205) and Pnm (No. 224) structures have been inferred from RXS measurements.125
| Initial configuration | Relative energy (eV) | Band-gap (eV) | Magnetic moment (μB per Np ion) | Lattice volume (Å3) | Space group (number) | |
|---|---|---|---|---|---|---|
| Ferromagnetic | ||||||
| (001) | 0.081 | 2.65 | 2.63 | 158.74 | I4/mmm (No. 139) | |
| (011) | 0.017 | 2.65 | 2.63 | 158.85 | Immm (No. 71) | |
| (111) | 0.000 | 2.42 | 2.67 | 158.82 |
Rm (No. 166) |
|
| Antiferromagnetic | ||||||
| Longitudinal | 1k | 0.079 | 2.65 | 2.60 | 158.78 | I4/mmm (No. 139) |
| 2k | 0.083 | 3.14 | 2.52 | 158.69 | I4/mmm (No. 139) | |
| 3k | 0.004 | 2.88 | 2.64 | 158.69 |
Fmm (No. 225) |
|
| Transverse | 1k | 0.064 | 3.26 | 2.55 | 158.74 | Fmmm (No. 69) |
| 2k | 0.001 | 3.20 | 2.64 | 158.79 | Pbca (No. 61) | |
| 3k | 0.002 | 3.20 | 2.64 | 158.71 |
Pa (No. 205) |
|
| Experimental | ||||||
| — | 2.85–3.10126,127 | ∼0.01–0.1093,96 | 159.8496 |
Fmm (No. 225),96 |
||
|
Pa (No. 205)104,105 |
||||||
The HSE06 results above are emulated by PBEsol+U (Fig. 5). The FM (111) ground-state, calculated by PBEsol, independent of the U modifier, results in Rm (No. 166) symmetry as a result of a trigonal distortion of the unit cell. However, this structure is inconsistent with current experimental data. A number of experimental investigations have strongly indicated an AFM ground-state, although the nature of the magnetic moment has thus far not been determined. No evidence of a trigonal Rm (No. 166) crystallographic distortion has been reported, but noncollinear 3k AFM behaviour has been identified, where the domain is unresolved.96,97,105,125,128 From our calculations, only the noncollinear 3k AFM states result in cubic symmetry. The transverse 3k AFM state is marginally lower in energy than the longitudinal 3k AFM state (Table 4), with the external cubic symmetry retained in both cases.
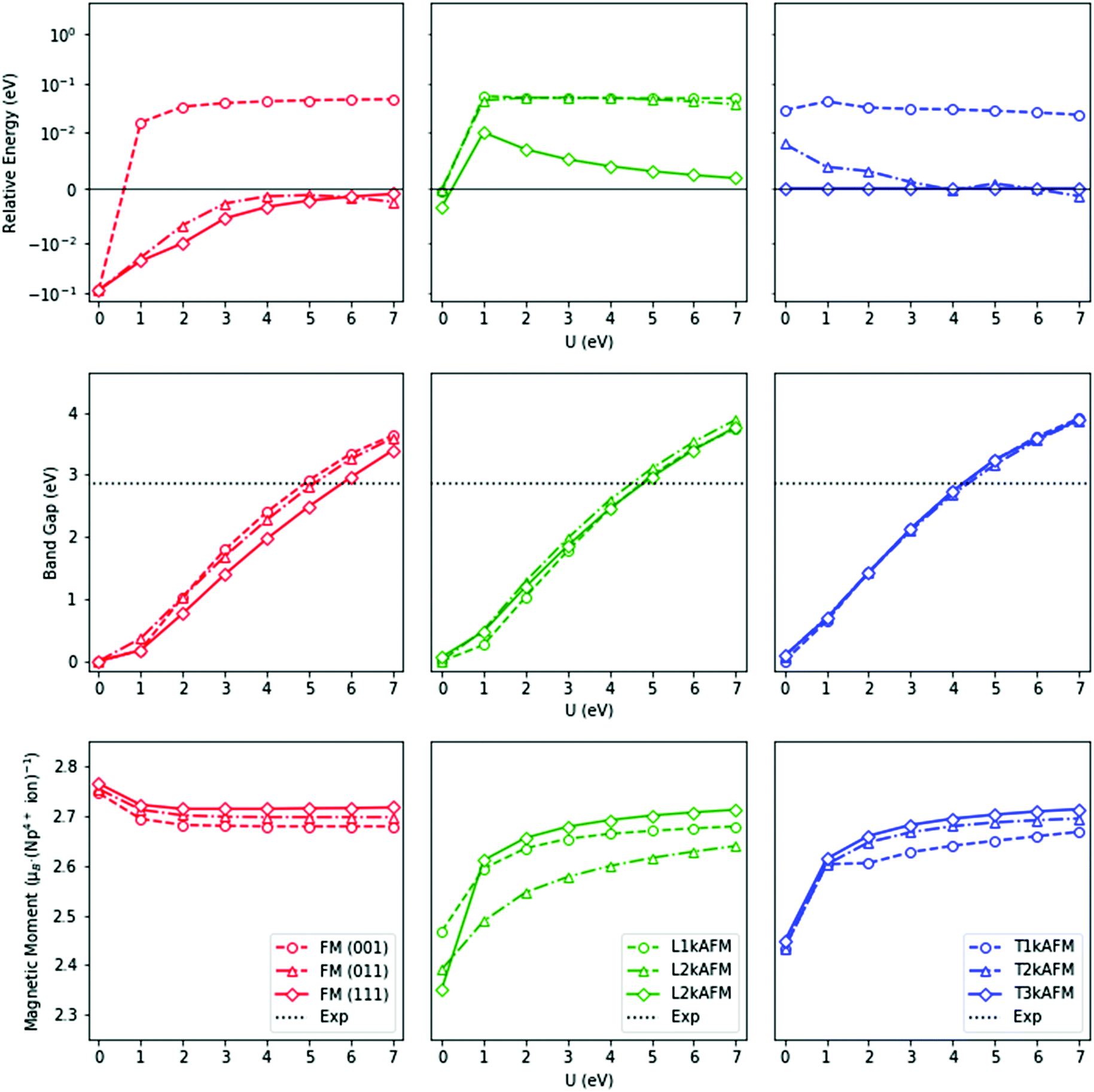 | ||
| Fig. 5 The relative ground-state energies, band-gaps, and effective magnetic moments against the Coulomb modifier (U) for ferromagnetic (FM), and antiferromagnetic (AFM) states of NpO2, calculated with PBEsol+U. The antiferromagnetic transverse (T) and longitudinal (L) domains are additionally represented. The k-prefix denotes the number of independent wave vectors: above, the calculated energy of magnetic states relative to the transverse 3k antiferromagnetic state; middle, the fundamental band-gap; below, the effective magnetic moment of the neptunium ions. The FM (orange), longitudinal AFM (green) and transverse AFM (blue) states are indicated. | ||
The electronic structure of NpO2 is influenced by the magnetic state. The measured optical absorbance band-gap of NpO2 epitaxial thin films is 2.85 eV.126 In our calculations, the correct insulating nature of NpO2 is reproduced when U = 4–6 eV for all magnetic states. In this range, the relative energy differences between the longitudinal 3k AFM, transverse 2–3k AFM, and the FM (111) states are minimal.
The longitudinal 3k AFM state results in Fmm (No. 225) symmetry, which is supported by RXS measurements. In contrast, the transverse 3k AFM state results in Pa (No. 205) symmetry, characterized by an internal O2− distortion of 0.011 Å. The distortion is equal to that implied by Mossbauer spectroscopy93 and INS104,105 measurements. In theory, the noncollinear AFM domain can be established by its crystalline environment, but the distortion cannot be confirmed as this is below the instrument resolution. In terms of the lattice volume, no discernible change between different magnetic states is observed.
As mentioned above, the magnetic moment of the Np ions is unresolved. Experimentally, the NpO2 system appears to be a small-moment system, but this picture cannot be confirmed by first-principle methods. The magnetic moment of the Np ions in the FM states decreases from 2.77 μB per Np ion to 2.68 μB per Np ion when U ranges from 0–7 eV, whereas in AFM states, it increases from 2.35 μB per Np ion to 2.71 μB per Np ion for the same values of U = 0–7 eV. In contrast with low-temperature experimental measurements, the calculated magnetic moment is considerably greater. A superexchange-type mechanism in NpO2 could result in a small-moment system, for which DFT-based methods have been shown to be unsuitable.19,22,129 It is noted that in reference to the high-temperature PM state, the calculated magnetic moment is in close agreement.
crystal symmetry in UO2.13 We therefore employed the same approach used for UO2, whereby the electronic structure of the transverse 3k AFM (U = 4.25 eV) has been calculated by the PBEsol functional (Fig. 6). The electronic structure of the longitudinal 3k AFM state with Fmm crystal symmetry has been shown for comparison; here, the U (5.00 eV) and J (0.75 eV) values are taken from an earlier investigation.33,48
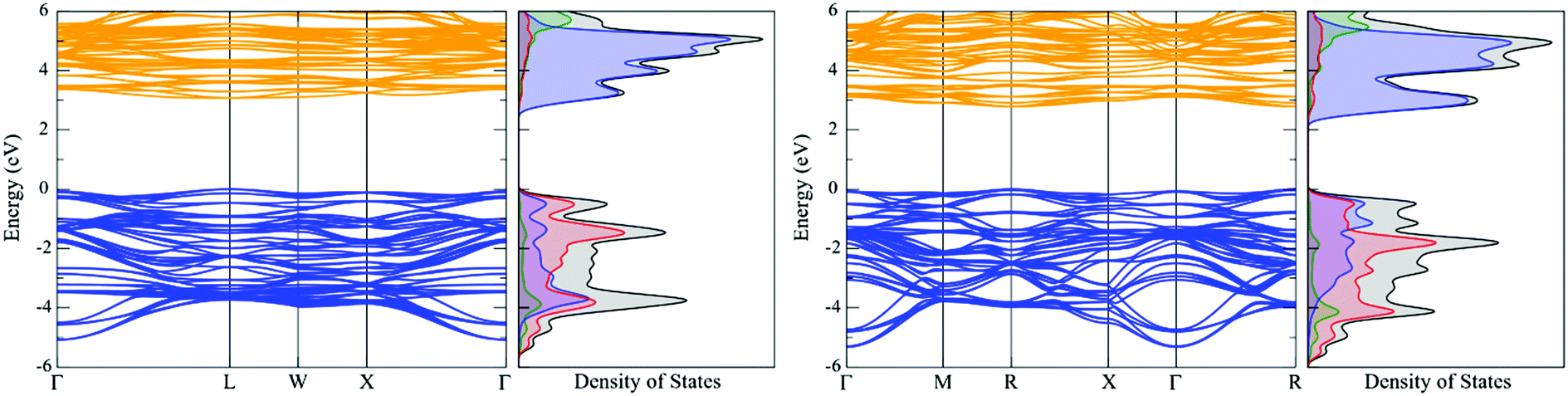 | ||
| Fig. 6 The electronic structure of NpO2 calculated by PBEsol for the: (left) longitudinal 3k AFM state (U = 5.00 eV, J = 0.75 eV); (right) transverse 3k AFM state (U = 4.25 eV, J = 0.00 eV). The valence band (dark blue), conduction band (orange), Np f- (blue), Np d- (green) and O p- (red) states are indicated. | ||
In the transverse 3k AFM state, the CMB is formed equally of oxygen p-states and neptunium f-states, indicating that NpO2 shares both Mott and charge-transfer characteristics. In contrast, the longitudinal 3k AFM state predominately results in a charge-transfer insulator.33 The charge-transfer characteristics are likely due to the absence of an exchange modifier that otherwise serves to reduce the effective Np magnetic moment. In both instances, the Np d-states have no significant role in chemical bonding interactions. The band structure reveals a direct R-centred band-gap of 2.79 eV, which compares with a calculated optical absorbance of 2.81 eV. Finally, the calculated bulk modulus of NpO2 for the longitudinal 3k AFM state is 214 GPa, whereas the bulk modulus for the transverse 3k AFM state is 215 GPa (Table 5).
| DFT+U | Band-gap (eV) | Lattice constant (Å) | Bulk modulus (GPa) | Magnetic moment (μB per Np ion) | Crystal symmetry | Magnetic state | ||
|---|---|---|---|---|---|---|---|---|
| U (eV) | J (eV) | Fundamental | Optical | |||||
| 4.25 | 0.00 | 2.79 | 2.81 | 5.442 | 215 | 2.70 |
Pa (205) |
T-3k AFM |
| 5.00 | 0.75 | 3.08 | 3.11 | 5.448 | 214 | 1.87 |
Fmm (225) |
L-3k AFM33 |
| — | — | 2.85–3.10126,127 | 5.42796 | 20011 | ∼0.01–0.1093,96 |
Fmm (225), Pa (205) |
Experimental | |
4. Conclusions
The magnetic structure of UO2 and NpO2 has been investigated by first-principles methods. The influence of the magnetic structure on crystal symmetry is considered, and has been observed in other systems.1 The direction and magnitude of the ionic magnetic forces in the AnO2 introduce stresses within the crystal, which influences the low-temperature structure. In our calculations, the crystal structure and magnetic environment are closely interlinked. The experimental cubic environment is only preserved by noncollinear 3k AFM states. The longitudinal 3k AFM state results in Fmm (No. 225) crystal symmetry, whereas the energetically marginally more favourable transverse 3k AFM state results in Pa (No. 205) crystal symmetry, with a distortion of the O2− ions of 0.011 Å. In contrast, the collinear 1k AFM states, often employed in past investigations, result in either an orthorhombic Fmmm (No. 69) or tetragonal I4/mmm (No. 139) distortion.131
A degenerate transverse 1–2k AFM ground-state for UO2 has been calculated, where the transverse 1k AFM state is in agreement with both static and low-frequency measurements of spin-wave excitations.22 The result contradicts the transverse 3k AFM state with Pa (No. 205) crystal symmetry found by: INS,77 RXS, and NMR measurements.20,77 As the ordered magnetic states are in close energetic proximity (<0.026 eV F.U.−1), thermal fluctuations are thought to play a significant role and the influence of entropy on the dynamic stability remains unknown. In addition, a FM (111) NpO2 ground-state with Rm (No. 166) has been found. This contradicts resonant X-ray scattering73 and 17O NMR measurements.97 In contrast, the transverse 3k AFM state with Pa (No. 205) crystal symmetry is only 0.001 eV higher in energy. This magnetic structure has been linked to experimental measurements.96,98,105,128,130 The insulating nature of NpO2 is reproduced by PBEsol+U when U = 4–6 eV for all magnetic configurations. In this study, the existence of a small-moment system cannot be confirmed.22,106,107
As the magnetic ground-sate from HSE06 calculations can be realised by PBEsol+U (where an appropriate U value has been chosen), PBEsol+U offers a means of modelling the electronic structure in larger systems.132 To keep the methodology concurrent with the commonly used Dudarev et al. formulism, the choice of U for transverse 1k AFM UO2 (3.50 eV) and transverse 3k AFM NpO2 (4.25 eV) where J = 0.00 eV has been shown. To avoid the crystallographic distortion of cubic symmetry found in the transverse 1k AFM UO2 state, transverse 3k AFM (U = 3.35 eV, J = 0.00 eV) order can also be used to emulate the electronic structure.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the UK Engineering & Physical Science Research Council (EPSRC) (Grant no. EP/G036675 and EP/K016288) and the Atomic Weapons Establishment (AWE). AES gratefully acknowledges the United States Department of Homeland Security (DHS), Domestic Nuclear Detection Office (DNDO), National Technical Nuclear Forensics Centre (NTNFC), for a Postdoctoral Research Fellowship. NHdL thanks AWE for a William Penney Fellowship. This work made use of the ARCHER UK National Supercomputing Service (http://www.archer.ac.uk), via our membership of the UK's HEC Materials Chemistry Consortium, which is funded by EPSRC (EP/L000202).Notes and references
- J. T. Pegg, A. E. Shields, M. T. Storr, A. S. Wills, D. O. Scanlon and N. H. de Leeuw, Phys. Chem. Chem. Phys., 2018, 20, 20943–20951 RSC.
- J. Schoenes, Phys. Rep., 1980, 63, 301–336 CrossRef.
- G. Kalkowski, G. Kaindl, W. D. Brewer and W. Krone, Phys. Rev. B: Condens. Matter Mater. Phys., 1987, 35, 2667–2677 CrossRef CAS.
- M. Noe and J. Fuger, Inorg. Nucl. Chem. Lett., 1974, 10, 7–19 CrossRef CAS.
- A. Arrott and J. E. Goldman, Phys. Rev., 1957, 108, 948–953 CrossRef CAS.
- J. K. Dawson and M. W. Lister, J. Chem. Soc., 1950, 2181–2187, 10.1039/JR9500002181.
- C. E. McNeilly, J. Nucl. Mater., 1964, 11, 53–58 CrossRef CAS.
- T. N. Taylor and W. P. Ellis, Surf. Sci., 1981, 107, 249–262 CrossRef CAS.
- I. D. Prodan, G. E. Scuseria, J. A. Sordo, K. N. Kudin and R. L. Martin, J. Chem. Phys., 2005, 123, 014703 CrossRef.
- Y. Tokunaga, H. Sakai, T. Fujimoto, S. Kambe, R. E. Walstedt, K. Ikushima, H. Yasuoka, D. Aoki, Y. Homma, Y. Haga, T. D. Matsuda, S. Ikeda, E. Yamamoto, A. Nakamura, Y. Shiokawa, K. Nakajima, Y. Arai and Y. Ōnuki, J. Alloys Compd., 2007, 444–445, 241–245 CrossRef CAS.
- M. Idiri, T. Le Bihan, S. Heathman and J. Rebizant, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 70, 014113 CrossRef.
- S. D. Conradson, B. D. Begg, D. L. Clark, C. den Auwer, M. Ding, P. K. Dorhout, F. J. Espinosa-Faller, P. L. Gordon, R. G. Haire, N. J. Hess, R. F. Hess, D. Webster Keogh, G. H. Lander, D. Manara, L. A. Morales, M. P. Neu, P. Paviet-Hartmann, J. Rebizant, V. V. Rondinella, W. Runde, C. Drew Tait, D. Kirk Veirs, P. M. Villella and F. Wastin, J. Solid State Chem., 2005, 178, 521–535 CrossRef CAS.
- L. Desgranges, Y. Ma, P. Garcia, G. Baldinozzi, D. Siméone and H. E. Fischer, Inorg. Chem., 2016, 56, 321–326 CrossRef PubMed.
- S. Steiner, S. Khmelevskyi, M. Marsmann and G. Kresse, Phys. Rev. B: Condens. Matter Mater. Phys., 2016, 93, 224425 CrossRef.
- S. J. Allen, Phys. Rev., 1968, 166, 530–539 CrossRef CAS.
- S. J. Allen, Phys. Rev., 1968, 167, 492–496 CrossRef CAS.
- V. S. Mironov, L. F. Chibotaru and A. Ceulemans, Advances in Quantum Chemistry, Academic Press, 2003, vol. 44, pp. 599–616 Search PubMed.
- P. Giannozzi and P. Erdös, J. Magn. Magn. Mater., 1987, 67, 75–87 CrossRef CAS.
- P. Santini, S. Carretta, G. Amoretti, R. Caciuffo, N. Magnani and G. H. Lander, Rev. Mod. Phys., 2009, 81, 807–863 CrossRef CAS.
- K. Ikushima, S. Tsutsui, Y. Haga, H. Yasuoka, R. E. Walstedt, N. M. Masaki, A. Nakamura, S. Nasu and Y. Ōnuki, Phys. Rev. B: Condens. Matter Mater. Phys., 2001, 63, 104404 CrossRef.
- T. Sabine, G. Smith and K. Reeve, J. Phys. C: Solid State Phys., 1974, 7, 4513 CrossRef CAS.
- P. Santini, R. Lémanski and P. Erdős, Adv. Phys., 1999, 48, 537–653 CrossRef CAS.
- J. M. Haschke, J. Alloys Compd., 1998, 278, 149–160 CrossRef CAS.
- J. M. Haschke and T. H. Allen, J. Alloys Compd., 2001, 320, 58–71 CrossRef CAS.
- J. M. Haschke, T. H. Allen and J. C. Martz, J. Alloys Compd., 1998, 271–273, 211–215 CrossRef CAS.
- J. M. Haschke, T. H. Allen and L. A. Morales, J. Alloys Compd., 2001, 314, 78–91 CrossRef CAS.
- J. M. Haschke, T. H. Allen and L. A. Morales, Los Alamos Sci., 2000, 26 Search PubMed.
- J. M. Haschke, T. H. Allen and J. L. Stakebake, J. Alloys Compd., 1996, 243, 23–35 CrossRef CAS.
- J. M. Haschke and J. C. Martz, Los Alamos Sci., 2000, 26 Search PubMed.
- D. W. Osborne and E. F. W. Jr., J. Chem. Phys., 1953, 21, 1884–1887 CrossRef CAS.
- P. Hohenberg and W. Kohn, Phys. Rev., 1964, 136, B864–B871 CrossRef.
- W. Kohn and L. J. Sham, Phys. Rev., 1965, 140, A1133–A1138 CrossRef.
- J. T. Pegg, X. Aparicio-Anglès, M. Storr and N. H. de Leeuw, J. Nucl. Mater., 2017, 492, 269–278 CrossRef CAS.
- H. Wang and K. Konashi, J. Alloys Compd., 2012, 533, 53–57 CrossRef CAS.
- J. P. Perdew and A. Zunger, Phys. Rev. B: Condens. Matter Mater. Phys., 1981, 23, 5048–5079 CrossRef CAS.
- S. L. Dudarev, G. A. Botton, S. Y. Savrasov, C. J. Humphreys and A. P. Sutton, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 57, 1505–1509 CrossRef CAS.
- A. I. Liechtenstein, V. I. Anisimov and J. Zaanen, Phys. Rev. B: Condens. Matter Mater. Phys., 1995, 52, R5467–R5470 CrossRef CAS.
- V. I. Anisimov, J. Zaanen and O. K. Andersen, Phys. Rev. B: Condens. Matter Mater. Phys., 1991, 44, 943–954 CrossRef CAS.
- A. Georges, G. Kotliar, W. Krauth and M. J. Rozenberg, Rev. Mod. Phys., 1996, 68, 13 CrossRef CAS.
- C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158–6170 CrossRef CAS.
- J. Heyd, G. E. Scuseria and M. Ernzerhof, J. Chem. Phys., 2003, 118, 8207–8215 CrossRef CAS.
- I. D. Prodan, G. E. Scuseria and R. L. Martin, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 76, 033101 CrossRef.
- M.-T. Suzuki, H. Ikeda and P. M. Oppeneer, J. Phys. Soc. Jpn., 2018, 87, 041008 CrossRef.
- X.-D. Wen, R. L. Martin, L. E. Roy, G. E. Scuseria, S. P. Rudin, E. R. Batista, T. M. McCleskey, B. L. Scott, E. Bauer and J. J. Joyce, J. Chem. Phys., 2012, 137, 154707 CrossRef PubMed.
- J. Kolorenč, A. B. Shick and A. I. Lichtenstein, Phys. Rev. B: Condens. Matter Mater. Phys., 2015, 92, 085125 CrossRef.
- Q. Yin, A. Kutepov, K. Haule, G. Kotliar, S. Y. Savrasov and W. E. Pickett, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 84, 195111 CrossRef.
- Q. Yin and S. Y. Savrasov, Phys. Rev. Lett., 2008, 100, 225504 CrossRef PubMed.
- M. T. Suzuki, N. Magnani and P. M. Oppeneer, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 88, 195146 CrossRef.
- D. Gryaznov, E. Heifets and D. Sedmidubsky, Phys. Chem. Chem. Phys., 2010, 12, 12273–12278 RSC.
- P. S. Ghosh, N. Kuganathan, A. Arya and R. W. Grimes, Phys. Chem. Chem. Phys., 2018, 20, 18707–18717 RSC.
- X.-D. Wen, R. L. Martin, G. E. Scuseria, S. P. Rudin and E. R. Batista, J. Phys. Chem. C, 2013, 117, 13122–13128 CrossRef CAS.
- I. D. Prodan, G. E. Scuseria and R. L. Martin, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 73, 045104 CrossRef.
- D. A. Andersson, J. Lezama, B. P. Uberuaga, C. Deo and S. D. Conradson, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 79, 024110 CrossRef.
- Y. Yang, B. Wang and P. Zhang, J. Nucl. Mater., 2013, 433, 345–350 CrossRef CAS.
- B. Sun, H. Liu, H. Song, G. Zhang, H. Zheng, X. Zhao and P. Zhang, J. Nucl. Mater., 2012, 426, 139–147 CrossRef CAS.
- P. Zhang, B.-T. Wang and X.-G. Zhao, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 144110 CrossRef.
- D. Gryaznov, S. Rashkeev, E. Kotomin, E. Heifets and Y. Zhukovskii, Nucl. Instrum. Methods Phys. Res., Sect. B, 2010, 268, 3090–3094 CrossRef CAS.
- G. Jomard, B. Amadon, F. Bottin and M. Torrent, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 78, 075125 CrossRef.
- G. Jomard and F. Bottin, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 84, 195469 CrossRef.
- X.-D. Wen, R. L. Martin, T. M. Henderson and G. E. Scuseria, Chem. Rev., 2013, 113, 1063–1096 CrossRef CAS PubMed.
- J. Boettger and A. Ray, Int. J. Quantum Chem., 2002, 90, 1470–1477 CrossRef CAS.
- S. A. Moten, R. Atta-Fynn, A. K. Ray and M. N. Huda, J. Nucl. Mater., 2016, 468, 37–45 CrossRef CAS.
- B. Dorado and P. Garcia, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 87, 195139 CrossRef.
- A. Shick, J. Kolorenč, L. Havela, T. Gouder and R. Caciuffo, Phys. Rev. B: Condens. Matter Mater. Phys., 2014, 89, 041109 CrossRef.
- J. B. Comly, J. Appl. Phys., 1968, 39, 716–718 CrossRef CAS.
- R. Caciuffo, G. Amoretti, P. Santini, G. H. Lander, J. Kulda and P. D. V. Du Plessis, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 13892–13900 CrossRef CAS.
- W. M. Jones and E. A. L. Joseph Gordon, J. Chem. Phys., 1952, 20 Search PubMed.
- J. J. Huntzicker and E. F. Westrum, J. Chem. Thermodyn., 1971, 3, 61–76 CrossRef CAS.
- B. Frazer, G. Shirane, D. Cox and C. Olsen, Phys. Rev., 1965, 140, A1448 CrossRef.
- B. Frazer, G. Shirane, D. Cox and C. Olsen, J. Appl. Phys., 1966, 37, 1386 CrossRef.
- J. Faber, G. H. Lander and B. R. Cooper, Phys. Rev. Lett., 1975, 35, 1770–1773 CrossRef CAS.
- S. B. Wilkins, R. Caciuffo, C. Detlefs, J. Rebizant, E. Colineau, F. Wastin and G. H. Lander, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 73, 060406 CrossRef.
- S. B. Wilkins, J. A. Paixão, R. Caciuffo, P. Javorsky, F. Wastin, J. Rebizant, C. Detlefs, N. Bernhoeft, P. Santini and G. H. Lander, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 70, 214402 CrossRef.
- J. Faber and G. H. Lander, Phys. Rev. B: Solid State, 1976, 14, 1151–1164 CrossRef CAS.
- O. G. Brandt and C. T. Walker, Phys. Rev., 1968, 170, 528–541 CrossRef CAS.
- B. Dorado, G. Jomard, M. Freyss and M. Bertolus, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 035114 CrossRef.
- E. Blackburn, R. Caciuffo, N. Magnani, P. Santini, P. J. Brown, M. Enderle and G. H. Lander, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 72, 184411 CrossRef.
- S. W. Yu, J. G. Tobin, J. C. Crowhurst, S. Sharma, J. K. Dewhurst, P. Olalde-Velasco, W. L. Yang and W. J. Siekhaus, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 83, 165102 CrossRef.
- J. Schoenes, J. Appl. Phys., 1978, 49, 1463–1465 CrossRef CAS.
- F. Jollet, T. Petit, S. Gota, N. Thromat, M. Gautier-Soyer and A. Pasturel, J. Phys.: Condens. Matter, 1997, 9, 9393 CrossRef CAS.
- B. W. Veal and D. J. Lam, Phys. Lett. A, 1974, 49, 466–468 CrossRef.
- B. W. Veal and D. J. Lam, Phys. Rev. B: Solid State, 1974, 10, 4902–4908 CrossRef CAS.
- Y. Baer and J. Schoenes, Solid State Commun., 1980, 33, 885–888 CrossRef CAS.
- Y. Baer, Physica B+C, 1980, 102, 104–110 CrossRef CAS.
- L. E. Cox, W. P. Ellis, R. D. Cowan, J. W. Allen, S. J. Oh, I. Lindau, B. B. Pate and A. J. Arko, Phys. Rev. B: Condens. Matter Mater. Phys., 1987, 35, 5761–5765 CrossRef CAS.
- P. Roussel, P. Morrall and S. J. Tull, J. Nucl. Mater., 2009, 385, 53–56 CrossRef CAS.
- J. C. Boettger and A. K. Ray, Int. J. Quantum Chem., 2000, 80, 824–830 CrossRef CAS.
- K. N. Kudin, G. E. Scuseria and R. L. Martin, Phys. Rev. Lett., 2002, 89, 266402 CrossRef.
- T. Petit, G. Jomard, C. Lemaignan, B. Bigot and A. Pasturel, J. Nucl. Mater., 1999, 275, 119–123 CrossRef CAS.
- B. Ao, R. Qiu, H. Lu and P. Chen, J. Phys. Chem. C, 2016, 120, 18445–18451 CrossRef CAS.
- M. Blume, Phys. Rev., 1966, 141, 517–524 CrossRef CAS.
- M. R. Daniel, Phys. Lett., 1966, 22, 131–132 CrossRef CAS.
- J. M. Friedt, F. J. Litterst and J. Rebizant, Phys. Rev. B: Condens. Matter Mater. Phys., 1985, 32, 257–263 CrossRef CAS.
- P. Erdös, G. Solt, G. Zołnierek, A. Blaise and J. M. Fournier, Physica B+C, 1980, 102, 164–170 CrossRef.
- E. F. W. Jr., J. B. Hatcher and D. W. Osborne, J. Chem. Phys., 1953, 21, 419–423 CrossRef.
- D. Mannix, G. H. Lander, J. Rebizant, R. Caciuffo, N. Bernhoeft, E. Lidström and C. Vettier, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 60, 15187–15193 CrossRef CAS.
- Y. Tokunaga, Y. Homma, S. Kambe, D. Aoki, H. Sakai, E. Yamamoto, A. Nakamura, Y. Shiokawa, R. E. Walstedt and H. Yasuoka, Phys. Rev. Lett., 2005, 94, 137209 CrossRef CAS.
- R. Caciuffo, G. H. Lander, J. C. Spirlet, J. M. Fournier and W. F. Kuhs, Solid State Commun., 1987, 64, 149–152 CrossRef CAS.
- W. Kopmann, F. J. Litterst, H. H. Klauß, M. Hillberg, W. Wagener, G. M. Kalvius, E. Schreier, F. J. Burghart, J. Rebizant and G. H. Lander, J. Alloys Compd., 1998, 271–273, 463–466 CrossRef CAS.
- P. W. Anderson, Phys. Rev., 1950, 79, 350–356 CrossRef.
- W. J. De Haas, E. C. Wiersma and H. A. Kramers, Physica, 1934, 1, 1–13 CrossRef.
- C. B. Finch and G. W. Clark, J. Cryst. Growth, 1970, 6, 245–248 CrossRef CAS.
- J. C. Spirlet, E. Bednarczyk, C. Rijkeboer, C. Rizzoli, J. Rebizant and O. Vogt, Inorg. Chim. Acta, 1984, 94, 111–112 CrossRef.
- G. Amoretti, A. Blaise, R. Caciuffo, D. Di Cola, J. Fournier, M. Hutchings, G. Lander, R. Osborn, A. Severing and A. Taylor, J. Phys.: Condens. Matter, 1992, 4, 3459 CrossRef CAS.
- R. Caciuffo, G. Amoretti, J. M. Fournier, A. Blaise, R. Osborn, A. D. Taylor, J. Larroque and M. T. Hutchings, Solid State Commun., 1991, 79, 197–200 CrossRef CAS.
- C. Wang, M. Norman, R. Albers, A. Boring, W. Pickett, H. Krakauer and N. Christensen, Phys. Rev. B: Condens. Matter Mater. Phys., 1987, 35, 7260 CrossRef CAS.
- J. A. Mydosh and P. M. Oppeneer, Philos. Mag., 2014, 94, 3642–3662 CrossRef CAS.
- P. Santini, G. Amoretti, R. Caciuffo, F. Bourdarot and B. Fåk, Phys. Rev. Lett., 2000, 85, 654–657 CrossRef CAS PubMed.
- M. Colarieti-Tosti, O. Eriksson, L. Nordström, J. Wills and M. S. S. Brooks, Phys. Rev. B: Condens. Matter Mater. Phys., 2002, 65, 195102 CrossRef.
- S. Kern, C. K. Loong, G. L. Goodman, B. Cort and G. H. Lander, J. Phys.: Condens. Matter, 1990, 2, 1933 CrossRef CAS.
- G. Raphael and R. Lallement, Solid State Commun., 1968, 6, 383–385 CrossRef CAS.
- H. Yasuoka, G. Koutroulakis, H. Chudo, S. Richmond, D. K. Veirs, A. I. Smith, E. D. Bauer, J. D. Thompson, G. D. Jarvinen and D. L. Clark, Science, 2012, 336, 901–904 CrossRef CAS PubMed.
- V. I. Anisimov, Strong coulomb correlations in electronic structure calculations, CRC Press, 2000 Search PubMed.
- W. H. Press, S. A. Teukolsky, W. T. Vetterling and B. P. Flannery, Numerical Recipes: The Art of Scientific Computing, Cambridge University Press, 3rd edn, 2007 Search PubMed.
- K. Momma and F. Izumi, J. Appl. Crystallogr., 2011, 44, 1272–1276 CrossRef CAS.
- A. V. Krukau, O. A. Vydrov, A. F. Izmaylov and G. E. Scuseria, J. Chem. Phys., 2006, 125, 224106 CrossRef PubMed.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Solid State, 1976, 13, 5188 CrossRef.
- E. Bousquet and N. Spaldin, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 220402 CrossRef.
- G. I. Csonka, J. P. Perdew, A. Ruzsinszky, P. H. Philipsen, S. Lebègue, J. Paier, O. A. Vydrov and J. G. Ángyán, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 79, 155107 CrossRef.
- P. E. Blöchl, O. Jepsen and O. K. Andersen, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 49, 16223 CrossRef.
- T. T. Meek, B. von Roedern, P. G. Clem and R. J. Hanrahan Jr, Mater. Lett., 2005, 59, 1085–1088 CrossRef CAS.
- B. Amadon, T. Applencourt and F. Bruneval, Phys. Rev. B: Condens. Matter Mater. Phys., 2014, 89, 125110 CrossRef.
- T. Bo, J.-H. Lan, C.-Z. Wang, Y.-L. Zhao, C.-H. He, Y.-J. Zhang, Z.-F. Chai and W.-Q. Shi, J. Phys. Chem. C, 2014, 118, 21935–21944 CrossRef CAS.
- T. Bo, J.-H. Lan, Y.-L. Zhao, Y.-J. Zhang, C.-H. He, Z.-F. Chai and W.-Q. Shi, J. Nucl. Mater., 2014, 454, 446–454 CrossRef CAS.
- J. A. Paixão, C. Detlefs, M. J. Longfield, R. Caciuffo, P. Santini, N. Bernhoeft, J. Rebizant and G. H. Lander, Phys. Rev. Lett., 2002, 89, 187202 CrossRef PubMed.
- T. M. McCleskey, E. Bauer, Q. Jia, A. K. Burrell, B. L. Scott, S. D. Conradson, A. Mueller, L. Roy, X. Wen and G. E. Scuseria, J. Appl. Phys., 2013, 113, 013515 CrossRef.
- C. Suzuki, T. Nishi, M. Nakada, M. Akabori, M. Hirata and Y. Kaji, J. Phys. Chem. Solids, 2012, 73, 209–216 CrossRef CAS.
- R. Caciuffo, J. A. Paixão, C. Detlefs, M. J. Longfield, P. Santini, N. Bernhoeft, J. Rebizant and G. H. Lander, J. Phys.: Condens. Matter, 2003, 15, S2287 CrossRef CAS.
- P. Santini and G. Amoretti, Phys. Rev. Lett., 2000, 85, 2188–2191 CrossRef CAS PubMed.
- S. W. Lovesey, E. Balcar, C. Detlefs, G. V. D. Laan, D. S. Sivia and U. Staub, J. Phys.: Condens. Matter, 2003, 15, 4511 CrossRef CAS.
- J. T. Pegg, A Noncollinear Relativistic Computational Study of the Actinide Dioxides and their Interaction with Hydrogen, UCL (University College London), 2018 Search PubMed.
- J. T. Pegg, A. E. Shields, M. T. Storr, D. O. Scanlon and N. H. de Leeuw, J. Phys. Chem. C, DOI:10.1021/acs.jpcc.8b07823.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8cp03581d |
| ‡ This manuscript has been co-authored by UT-Battelle, LLC, under contract DE-AC05-00OR22725 with the US Department of Energy (DOE). The US government retains and the publisher, by accepting the article for publication, acknowledges that the US government retains a nonexclusive, paid-up, irrevocable, worldwide license to publish or reproduce the published form of this manuscript, or allow others to do so, for US government purposes. DOE will provide public access to these results of federally sponsored research in accordance with the DOE Public Access Plan (http://energy.gov/downloads/doe-public-access-plan). |
| This journal is © the Owner Societies 2019 |