
Halogen bonding in 5-iodo-1-arylpyrazoles investigated in the solid state and predicted by solution 13C-NMR spectroscopy†
Marcel Mirel
Popa
 a,
Isabela Costinela
Man
a,
Constantin
Draghici
a,
Sergiu
Shova
b,
Mino R.
Caira
c,
Florea
Dumitrascu
*a and
Denisa
Dumitrescu
d
a,
Isabela Costinela
Man
a,
Constantin
Draghici
a,
Sergiu
Shova
b,
Mino R.
Caira
c,
Florea
Dumitrascu
*a and
Denisa
Dumitrescu
d
a“C.D. Nenitzescu” Center for Organic Chemistry, Romanian Academy, Spl. Independentei 202b, Bucharest, Romania. E-mail: fdumitra@yahoo.com
b“Petru Poni” Institute of Macromolecular Chemistry, Department of Inorganic Polymers, Romanian Academy, Aleea Grigore Ghica Voda 41A, 700487 Iasi, Romania
cDepartment of Chemistry, University of Cape Town, Rondebosch 7701, South Africa
dOvidius University Constanta, Faculty of Pharmacy, Str. Cpt. Av. Al. Serbanescu 6, Campus Corp C, Constanta 900470, Romania
First published on 18th October 2019
Abstract
X-ray crystallography revealed the presence of halogen bonding in the crystal supramolecular structure of three highly substituted 1-arylpyrazoles. However the compounds 1–3 present different halogen bonding motifs that feature C–I⋯N (1), C–I⋯O (2) and C–I⋯π (3) contacts respectively. The magnitudes of the σ-hole corresponding to the iodine atom in the 5-iodo-1-arylpyrazoles 1–3 were calculated by DFT methods and the importance of halogen bonding as a significant stabilizing force within the crystal lattice was evaluated. The halogen bonding of 1-aryl-5-iodopyrazoles with several Lewis bases (Et3N, pyridine, DABCO or DMSO) was investigated by 13C NMR spectroscopy in the solution phase to confirm the halogen bonding affinity of the iodine atom. The most suitable reporting atom for the formation of the halogen bond is C-5 of the pyrazole ring, which is directly bonded to the iodine atom. The C-5 atom is significantly deshielded by as much as 6–7 ppm upon interaction with the Lewis bases in solution revealing the strong halogen bonding character of the iodine atom attached to C-5 of the pyrazole ring.
Introduction
In recent years there has been an increasing interest in halogen bonds (XBs)1–10 as important directional forces in molecular recognition processes. These forces occur in supramolecular structures ranging from two simple molecules acting as halogen bond donor and halogen bond acceptor to the most complex systems that feature protein–ligand recognition or protein folding mediated halogen interactions.11,12Briefly, halogen bonding is defined2 as a molecular interaction between an electrophilic region associated with a halogen atom in a molecular entity and a nucleophilic region (Lewis base) in another or the same molecule (Fig. 1). Such an electrophilic region is now well established under the concept of the σ-hole.13
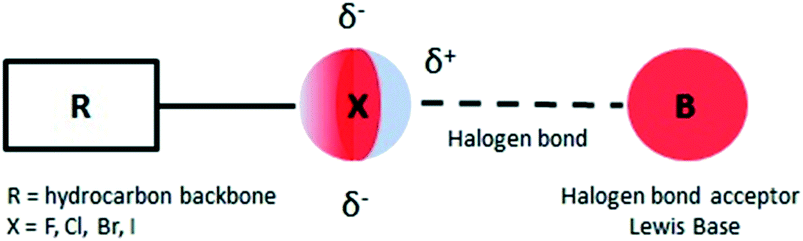 | ||
| Fig. 1 Schematic representation of the halogen bond. | ||
The propensity of two molecules to engage in halogen bonding is not as well understood as that for hydrogen bonding.1 The majority of published studies deal with halogen interactions in the solid state1–5 but the number of papers dealing with the study of halogen bonding in solution has increased in recent years.14–44
Halogenated pyrazoles were reported as synthons for studying the propensity of halogen bonding in different supramolecular architectures.45–49 Pyrazoles are important pharmaceutical lead compounds50 which, by halogenation, could increase their bioavailability owing to the remarkable interactions of halogen bonding donors with specific target enzymes.51
It appears that 5-iodopyrazoles were investigated to a lesser extent as halogen bond donors and searching the CCDC database we found some examples which present I⋯O, I⋯N or I⋯π contacts (CCDC refcodes: VEJPUY,48 ISODOK, ISODUQ, ISOFAY, ISOFEC, ISOFIG, ISOFOM, ISOFUS, ISOGAZ49) but the authors did not mention halogen bonding explicitly.
Herein we investigate the influence of the iodine bonding in three 5-iodopyrazoles on the supramolecular organization of the compounds in the solid state. X-ray diffraction analysis determined that the iodine atom of the 5-iodopyrazoles is involved in three main types of halogen bonding, namely C–I⋯N, C–I⋯O and C–I⋯π, which cannot be predicted from experiments performed in solution.
Results and discussion
Synthesis
The 5-iodopyrazoles under investigation (Fig. 2) were synthesized by 1,3-dipolar cycloaddition of the corresponding iodinated sydnones with dimethyl acetylenedicarboxylate (see ESI†).52,53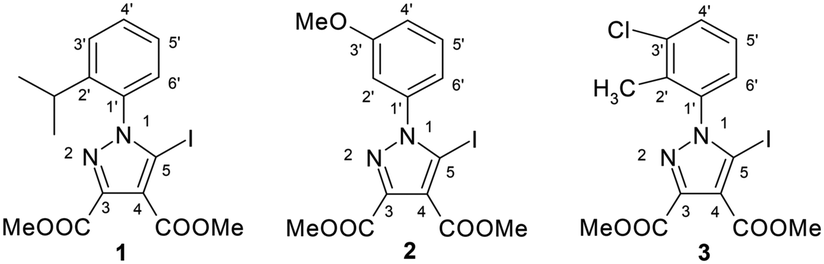 | ||
| Fig. 2 The structures of the dimethyl 1-aryl-5-iodopyrazole-3,4-dicarboxylates. | ||
Crystals suitable for further X-ray diffraction studies were obtained by slow evaporation of acetonitrile solution (1) and ethanol–methylene chloride solutions (2 and 3).
X-ray crystallography
According to single crystal X-ray diffraction all the studied compounds 1–3 have a supramolecular crystal structure built-up from neutral entities, as shown in Fig. 3. The crystallographic and geometrical parameters (bond lengths and angles) are summarized in Tables 1 and S1† respectively.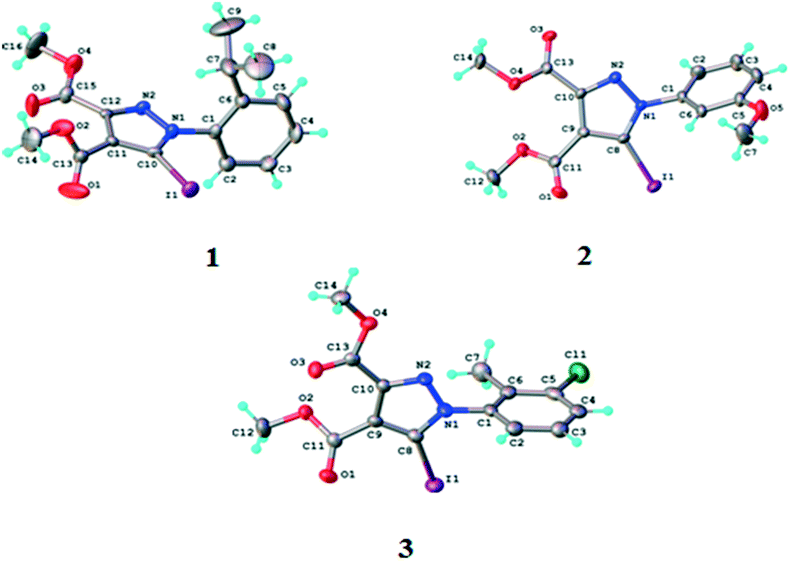 | ||
| Fig. 3 ORTEP diagrams of the molecules of compounds 1–3 with atom labelling schemes and thermal ellipsoids at the 40% probability level. | ||
| Compound | 1 | 2 | 3 |
|---|---|---|---|
| a R 1 = ∑||Fo| − |Fc||/∑|Fo|. b wR2 = {∑[w(Fo2 − Fc2)2]/∑[w(Fo2)2]}1/2. c GOF = {∑[w(Fo2 − Fc2)2]/(n − p)}1/2, where n is the number of reflections and p is the total number of parameters refined. | |||
| Formula | C16H17IN2O4 | C14H13IN2O5 | C14H12ClIN2O4 |
| CCDC no. | 1939541 | 1939542 | 1939543 |
| Fw [g mol−1] | 428.22 | 416.16 | 434.61 |
| Space group | P212121 | Cc |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
| a [Å] | 8.1400(6) | 23.0873(10) | 8.3915(7) |
| b [Å] | 13.4654(5) | 9.6374(3) | 10.0389(8) |
| c [Å] | 16.3082(8) | 7.2450(3) | 10.2106(9) |
| α [°] | 90.00 | 90.00 | 111.275(8) |
| β [°] | 90.00 | 106.961(4) | 96.662(7) |
| γ [°] | 90.00 | 90.00 | 95.904(7) |
| V [Å3] | 1787.52(16) | 1541.91(10) | 786.30(11) |
| Z | 4 | 4 | 2 |
| λ [Å] | MoKα 0.71073 | MoKα 0.71073 | MoKα 0.71073 |
| ρ calcd [g cm−3] | 1.591 | 1.793 | 1.836 |
| Crystal size [mm] | 0.30 × 0.15 × 0.15 | 0.20 × 0.15 × 0.15 | 0.30 × 0.25 × 0.25 |
| T [K] | 293 | 293 | 200 |
| μ [mm−1] | 1.811 | 2.102 | 2.225 |
| 2Θ range | 3.92–50.04 | 3.68–50.04 | 4.34–50.06 |
| Reflections collected | 5637 | 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 152 152 |
7393 |
| Independent reflections | 3139 [Rint = 0.0278] | 2731[Rint = 0.0369] | 2768[Rint = 0.0340] |
| Data/restraints/parameters | 3139/0/212 | 2731/2/202 | 2768/0/202 |
| R 1 | 0.0364 | 0.0257 | 0.0490 |
| wR2b | 0.0781 | 0.0460 | 0.1351 |
| GOFc | 1.048 | 1.023 | 1.053 |
| Flack parameter | −0.01(3) | −0.033(15) | — |
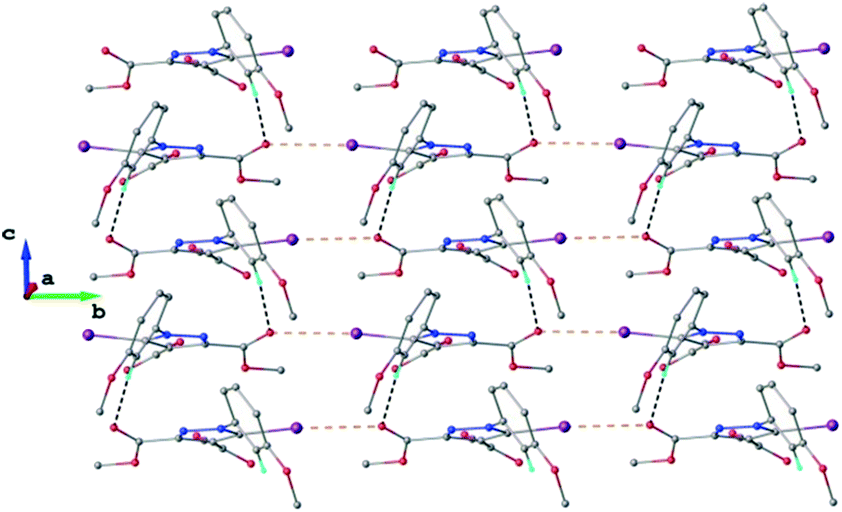
The crystal structure of 1 essentially results from the packing of 2D supramolecular wave-like layers propagated parallel to the (110) plane, as shown in Fig. 4. The analysis of these layers (Fig. S1†) shows the presence of intermolecular interactions of two types: a) CH⋯O hydrogen bonding and b) short I⋯N contacts at 2.992(4) Å, with C–I⋯N angle 174.4(1)°.
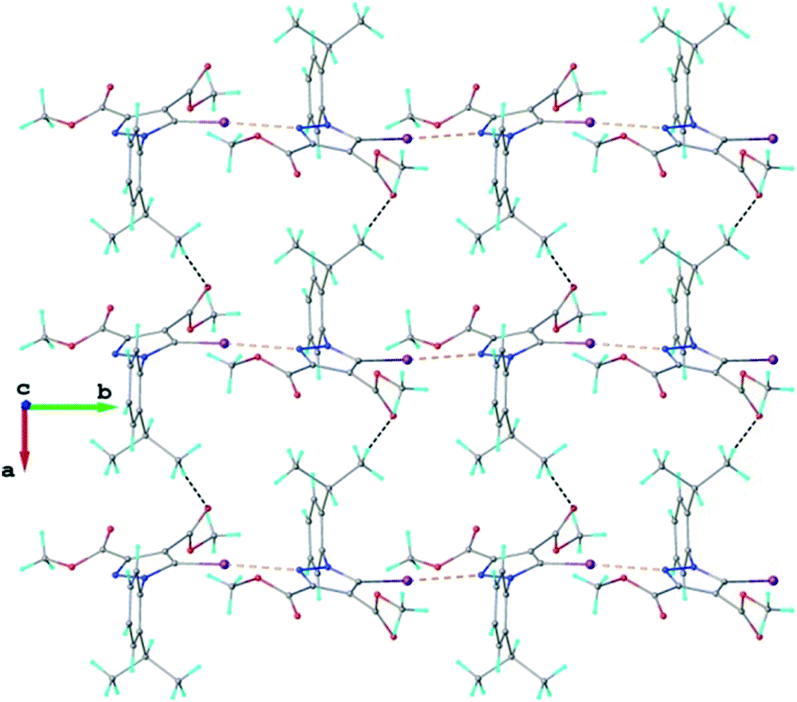 | ||
| Fig. 4 Two-dimensional supramolecular layer in the crystal structure of 1 viewed along the c-axis. The CH⋯O hydrogen bonds and the short I⋯N intermolecular contacts are shown in black and orange dashed lines, respectively. | ||
As found in compound 1, the main crystal structural motif in 2 can also be characterized as a 2D supramolecular network. As shown in Fig. 5, this architecture is stabilized by CH⋯O hydrogen bonding and the short contact between the iodine atom and the carbonyl oxygen atom (rather than the nitrogen atom, as occurs in 1). The short I⋯O contact distance is 3.099(3) Å and the C–I⋯O angle is 173.6(3)°. The parallel packing of isolated two-dimensional layers in the crystal structure of 2 is shown in Fig. S2.†
 | ||
| Fig. 5 Two-dimensional supramolecular layer in the crystal structure of 2 viewed along the a-axis. H-atoms not involved in hydrogen bonding are omitted for clarity. The CH⋯O hydrogen bonds and the short I⋯O intermolecular contacts are shown in black and orange dashed lines, respectively. | ||
In the crystal of 3 the neutral molecules interact to form a 2D supramolecular network, as shown in Fig. 6. The driving force in this case comprises C–H⋯O hydrogen bonding and a π–π-stacking interaction between inversion-related aromatic rings, as evidenced by the short centroid-to-centroid distance of 3.6971(3) Å (Fig. S3†).
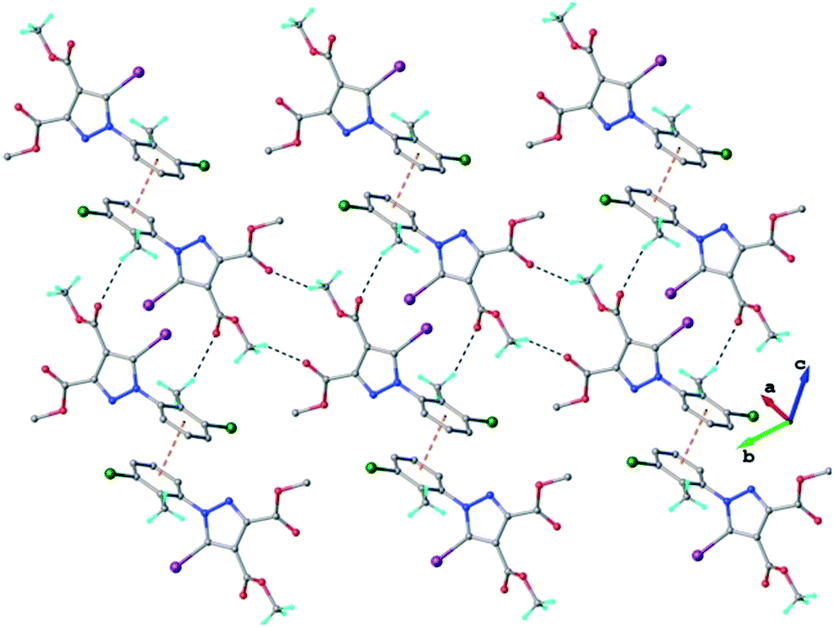 | ||
| Fig. 6 2D supramolecular network in the crystal structure of 3. The CH⋯O hydrogen bonds and the centroid-to-centroid distances are shown in black and orange dashed lines, respectively. | ||
However, a C–I⋯π type contact was observed along the C–I bond axis direction, which is perpendicular to the aryl ring plane (Fig. 7), and more precisely corresponding to a localized or above-the-atom C–I⋯π bond, with a distance between the iodine atom and C5′ atoms of 3.462 Å, which is less than the sum of their van der Waals (vdW) radii54 (3.689 Å) while the bonding angle C–I⋯C5′ is 173.4(2)°. These are typical geometrical characteristics of the π-type halogen bonding.
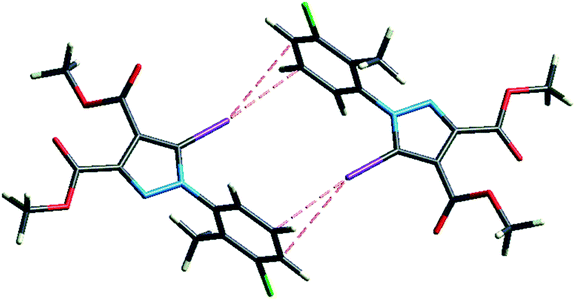 | ||
| Fig. 7 The C–I⋯π interaction showing iodine atom from one molecule directed perpendicular on the benzene ring from an adjacent molecule. | ||
Hirshfeld analysis
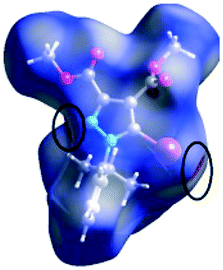
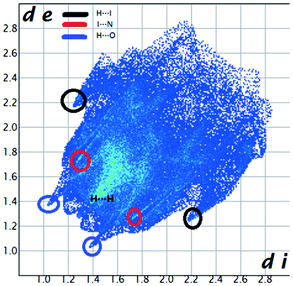
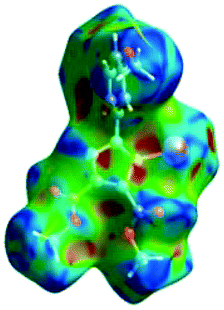
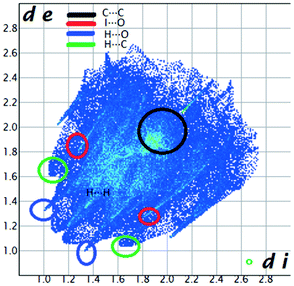
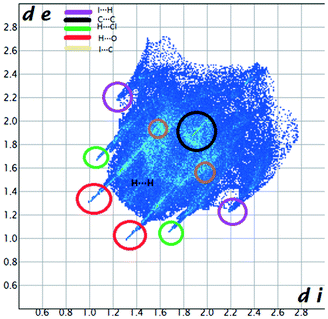
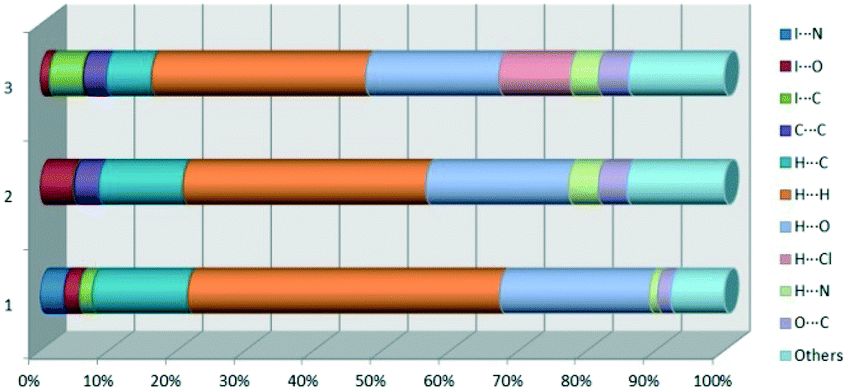
We investigated the intermolecular interactions by Hirshfeld analysis (as implemented in Crystal Explorer software)55 as one of the most descriptive ways to examine the contacts within a crystal structure. This is represented by the three-dimensional (3D) molecular Hirshfeld surfaces (HS) and the two- dimensional (2D) fingerprint plots of 3D HS, that simplify the complex information contained in the molecular crystal structure into a single plot which provides a “fingerprint” of the intermolecular interactions. The fingerprint plot of HS is calculated based on de, which is the distance from a point on the surface to the nearest nucleus outside the surface and di which is the distance from a point on the surface to the nearest nucleus inside the surface.56 The distance dnorm is the normalized contact distance, defined in terms of de, di and the vdW radii of the atoms. Distances shorter than vdW radii are marked in red spots, distances close to the vdW radii in white and distances larger than vdW in blue colour. The important interactions in 1–3 are summarized in the dnorm Hirshfeld surface (Tables 2 and S2†).| d norm | Shape index | Fingerprint plot | |
|---|---|---|---|
| 1 |
|
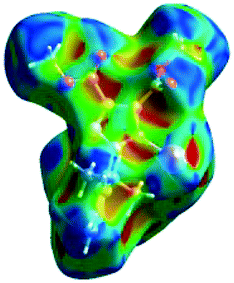
|
|
| 2 |
|
|
|
| 3 |
|

|
|
|
|||
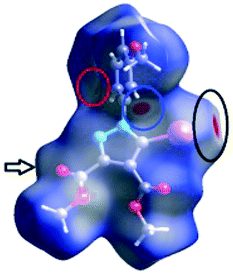
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond between the iodine and the oxygen atom in an ester moiety of the adjacent molecule which lead to the formation of infinite chains. The I⋯O contact distance is 3.09(9) Å which is 12% shorter than the sum of the vdW radii. The I⋯O contact was compared for over 500 compounds in the CCDC database (Fig. S5†) and found to fall in the range of strong I⋯O contacts. The infinite chains formed by the C–I⋯OC interaction are held together by CH⋯π interactions between the CH of the phenyl ring of one molecule and the phenyl ring of an adjacent molecule and by a C–H⋯OC hydrogen bond between the same pair of molecules, involving the H-5′ atom in the phenyl ring and an ester carbonyl oxygen atom. The Hirshfeld surface mapped with shape index presents one area of complementarity between two molecules (Fig. 8) corresponding to an interaction between two slightly offset pyrazole rings with centroid–centroid distances 3.74(9) Å. This interaction results in the formation of ribbon-like structures.
C bond between the iodine and the oxygen atom in an ester moiety of the adjacent molecule which lead to the formation of infinite chains. The I⋯O contact distance is 3.09(9) Å which is 12% shorter than the sum of the vdW radii. The I⋯O contact was compared for over 500 compounds in the CCDC database (Fig. S5†) and found to fall in the range of strong I⋯O contacts. The infinite chains formed by the C–I⋯OC interaction are held together by CH⋯π interactions between the CH of the phenyl ring of one molecule and the phenyl ring of an adjacent molecule and by a C–H⋯OC hydrogen bond between the same pair of molecules, involving the H-5′ atom in the phenyl ring and an ester carbonyl oxygen atom. The Hirshfeld surface mapped with shape index presents one area of complementarity between two molecules (Fig. 8) corresponding to an interaction between two slightly offset pyrazole rings with centroid–centroid distances 3.74(9) Å. This interaction results in the formation of ribbon-like structures.
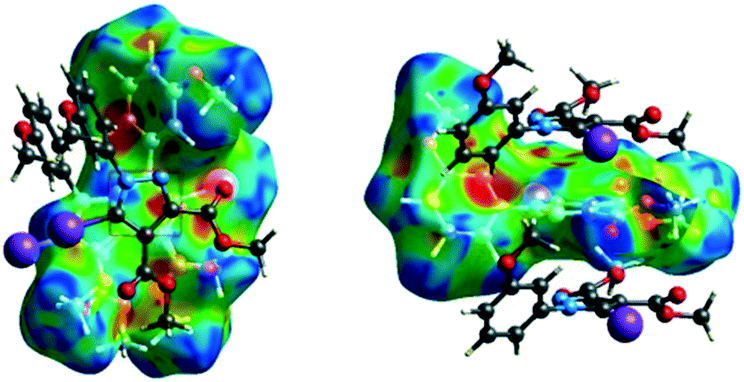 | ||
| Fig. 8 The shape index surface of compound 2 presenting the spatial arrangement of pyrazole rings to form a ribbon-like pattern. | ||
The fingerprint plot presents the main interactions in the crystal of 2, the most common being of type H⋯H accounting for 30% and also C⋯H and H⋯O interactions. Even though their frequency in the fingerprint plot is rather small as a percentage, the strongest interactions are I⋯O, I⋯H, O⋯H and π–π. Relative to 1 we can remark on the importance of C–H⋯π interactions in the crystal of 2 (see ESI† Table S2).
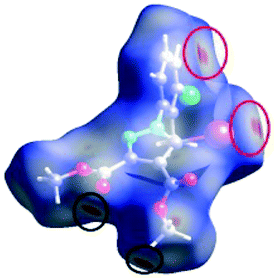
C between the methyl hydrogen in the ester moiety attached to C-4 of the pyrrole ring and the carbonyl oxygen in the ester attached to C-3 of the pyrrole ring of an adjacent symmetry-related molecule. The Shape Index surface of 3 presents two complementary regions (as small triangular patches of opposite colours red and blue).56 In Fig. 9a one can observe the existence of the π–π interaction between the phenyl rings of two molecules, previously discussed in the X-ray section as being an important stabilizing force of the primary 2D structure (Fig. 6). The shape index Hirshfeld surface of 3 shows also carbonyl-π short contacts between the CO group in the ester attached to C-3 and the pyrrole ring of an adjacent molecule while CO⋯OC short contacts appears between the carbonyl bond of the ester attached to C-3 and the carbonyl bond in the ester attached to C-3 of the same adjacent molecule, resulting a symmetric dimer structure (Fig. 9b).58,59 The fingerprint plot of 3 is the most complex, presenting strong H⋯O (19.5%), H⋯Cl (10.4%) and I⋯C (5%) interactions.
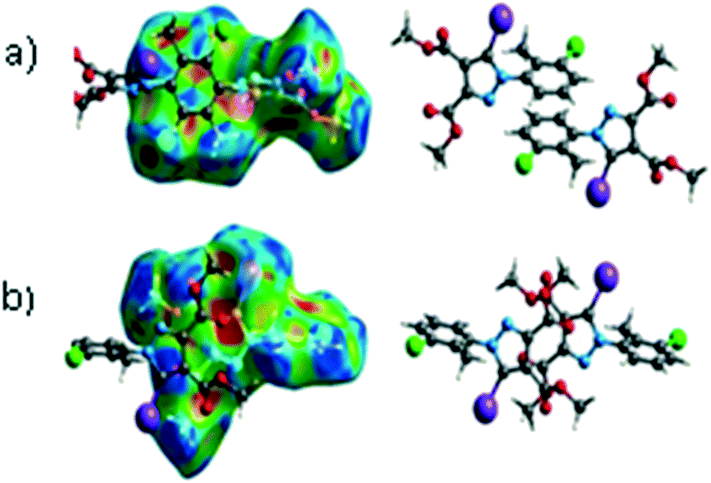 | ||
| Fig. 9 The presence of π⋯π (a) and CO⋯π (b) interactions predicted by the Hirshfeld surface (shape index). Coloured adjacent red and blue patches present regions of complementarity between two adjacent molecules revealing the presence of such interactions. | ||
At the end of Table 2 is presented a statistical representation of the main interactions in 1–3 (summarized as fingerprint plots by individual interactions in Table S2†) which qualitatively assesses the main driving forces which influence the crystal packing in the three compounds. The percentages of the halogen interactions reflect very well the type of halogen bonding in the three compounds. One can conclude that halogen bonds are important driving forces in stabilizing the supramolecular self-assembly, co-operating with other strong interactions such as hydrogen bonds.
The main X-ray interactions and features are summarized in Table 3:
| No. | D–X⋯A | d D–X [Å] | d X⋯A [Å] | <D–X⋯A [°] |
|---|---|---|---|---|
| 1 | C(10)–I(1)⋯N(2)(−x, 1/2 + y, 1/2 − z) | 2.059 | 2.992 | 174 |
| C(8)–H(8B)⋯O(1)C(1 + x, y, z) |
0.960 | 2.528 | 171 | |
| 2 | C(8)–I(1)⋯O(3)C(x, 1 + y, z) |
2.064 | 3.099 | 174 |
| C(6)–H(6)⋯O(3)C(x, 1 + y, z) |
0.930 | 2.491 | 168 | |
| 3 | C(12)–H(12C)⋯O(3)C(−x, −y, 1 − z) |
0.960 | 2.428 | 154 |
| C(7)–H(7B)⋯O(1)C(1 − x, 1 − y, 1 − z) |
0.959 | 2.594 | 156 | |
| C(12)–H(12A)⋯Cl(1)(−1 + x, 1 + y, 1 + z) | 0.960 | 2.859 | 147 | |
| C–I⋯ π | 2.055 | 3.430 | 165 |
Quantum mechanical calculations
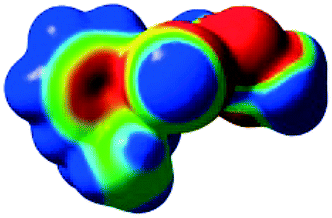
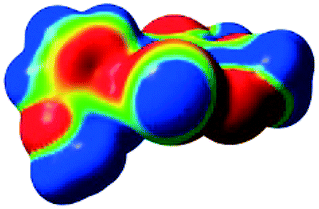
The characteristics of the supramolecular interactions in the three halogen bonds in compounds 1–3 were also investigated by quantum mechanical calculations in order to add a quantitative meaning to the halogen bond interactions and to rationalize their importance in the crystals among the other interactions. One of the most valuable concepts for the characterization of the halogen bonding is the “σ-hole” which was first introduced by Politzer et al. in 2007.13 The σ-hole defines an electropositive region centred along the axis of the R–X bond created when polarizable halogen atoms are bound to electronegative groups. Usually this region presents a positive electrostatic potential which permits the halogen atom to be engaged in attractive non-covalent interactions with different electronegative entities. The most important methods of calculation of the σ-hole parameters have been reviewed.60–62Quantum mechanical calculations were performed to measure the magnitude of the σ-holes of the iodine atoms in compounds 1–3. All calculations were performed with the G09 program suite63 using as starting point the crystalline structures obtained from single crystal X-ray diffraction. Because the positions of hydrogen atoms are not located accurately by X-ray diffraction, we have optimized their positions at the B3LYP-D3/dgdzvp level (B3LYP64,65 functional with the vdW dispersion correction66). The DGDZVP basis set is appropriate for all atomic species, without the need for any pseudopotential.67 The electrostatic potential map has been generated for the single molecules to gain insight into the nature and directionality of the halogen-bond. The electrostatic potential V(r) created by the electrons and nucleus of the molecule at any point r, has been proven to be an effective approach for interpreting and quantifying non-covalent interactions. For this purpose we compute V(r) on the molecular surface, which is defined as 0.001 e Bohr−3(a.u.) contour of the electronic density. This is a low electron density contour envelope that is in the magnitude range of the atomic vdW radii and was defined by Bader and is meaningful for non-covalent interactions.68 Electrostatic surface potentials were evaluated using the B3LYP-D3/def2tzvp basis set.69 The most positive values of the potentials at the halogen (local maximum) are referred to as Vs,max.61,62Tables 4 (and S4†) presents the ESP surfaces of molecules 1–3 rendered between −0.001 and +0.001 a.u. and the values of Vs,max of the σ-hole of the iodine atom (blue colour).
|
|
| V s,max = 110.2 kJ mol−11 | V s,max = 108.96 kJ mol−12 |
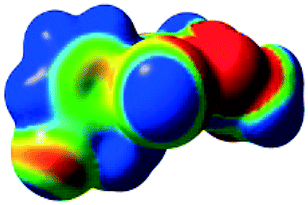
|

|
| V s,max= 118.15 kJ mol−13 | V s,max = 105.02 kJ mol−1 5-iodo-1H-pyrazole |
Compounds 1–3 present similar values for the σ-hole Vs,max magnitudes which could be characterized as medium to large, conferring on these compounds good XB donor capacity comparable with the σ-hole of some iodobenzimidazoles which form similar halogen bonding patterns.70 The values listed in Table 4 do not show much variation for the three compounds, suggesting that the different substituents on the phenyl ring of the pyrazole do not have a substantial influence. Furthermore, the 5-iodo-1H-pyrazole shown in Table 4 possesses a somewhat smaller value, but one that is within the range of the those for compounds 1–3. However, if the value of the σ-hole correlates somehow with the strength of the XB bonding one could not predict very reliably the electron-donating group which participates in halogen bond formation. This relies also on the presence of other non-covalent interactions occurring in the crystal structure. Certainly, the halogen bonding interaction is sufficiently strong to be considered a very important stabilizing force of the supramolecular assembly.
For comparative reasons, the binding energies of the dimers D1–D3 formed by halogen bonding between the iodine of one molecule and the corresponding Lewis base donor site of an adjacent molecule (Fig. 10) were calculated by DFT methods. Some of the other important interactions (i.e. HB, π⋯π stacking) were calculated in order to evaluate the relative contribution of the halogen bonding to the crystal structure (see ESI† for all the considered interactions).
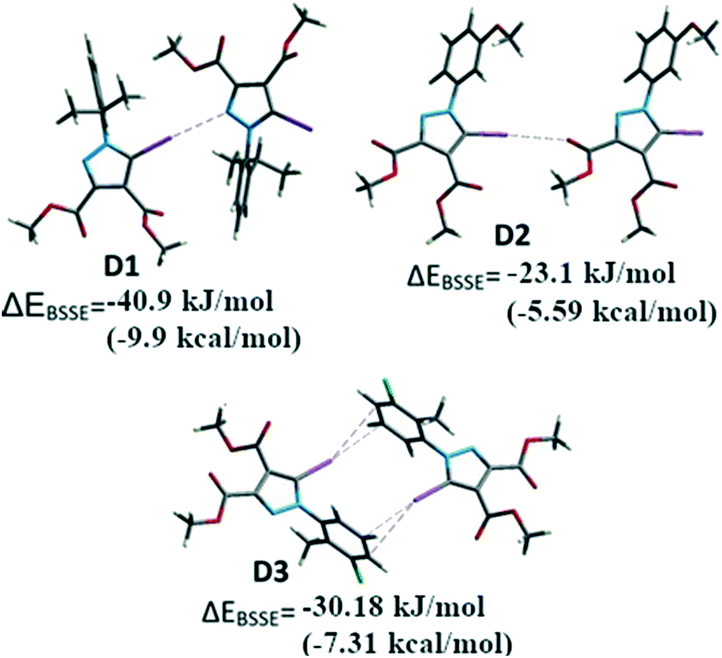 | ||
| Fig. 10 The dimers D1–D3 formed by I⋯N, I⋯O and I⋯π interactions respectively. | ||
Single point calculations for these dimeric units D1–D3 were performed using the B3LYP-D3 method combined with the DGDZVP basis set for iodine and 6-311++g(d,p) for other atoms, after the positions of the H atoms were optimized. The interaction energies71 corrected for BSSE (ΔEBSSE) of the dimers are presented in Fig. 13 for the halogen bonding interactions and in the Supplementary Information for the remaining important non-covalent interactions (Table S7†). The binding energy in D1 highlights a strong C–I⋯N interaction of −40.9 kJ mol−1 (−9.91 kcal mol−1) comparable with C–H⋯OC bonds which were calculated for D4 and D5 (Table S7†) of approximately −24.2 to −31.6 kJ mol−1 (−5.74 or −7.65 kcal mol−1). In a similar manner, for the compound 2 the dimer D2 presents the C–I⋯O interaction evaluated at approximately −23.1 kJ mol−1 (−5.6 kcal mol−1). However, the other important interactions such as the π⋯π stacking between the pyrazole rings could not be evaluated due to the fact that the energy values for D6 (see ESI,† Table S5) indicate the energy associated with the interplay of different interactions in the dimer that cannot be calculated separately by this method.
In some cases, the fragments comprise various, nearly identical interactions, related by symmetry elements in the crystal packing (D3, D7–D11-see ESI†). These interactions cannot be separated by the method employed. However, it appears for D3 the calculated energy is composed of the two symmetry-related C–I⋯π halogen bonds, suggesting a ΔEI⋯π of −15.1 kJ mol−1 (−3.61 kcal mol−1) (the halogen bond has relatively small interaction energy compared to the other halogen bonding presented above).
Halogen bonding in solution
Several methods of investigating the formation of halogen bonds in solution were developed, including NMR spectroscopy (1H,17,18,22,25,27,30,34–3913C,18–24,28,33,36,40,4119F,18,25,26,30,31,33,34,37,4015N,23,30,33,42 UV spectroscopy,41,43 IR spectroscopy22,26 and others.29,32,36 Most of the studies of XBs in solution relate to iodine as the most potent halogen bond donor, to perfluoro iodobenzenes with the fluorine atoms having a strong electron-withdrawing effect, thus strengthening the XB donor capacity, or to appropriate substituted ethynyl iodides.20,21,28,30,33Non-covalent interactions in the solution phase are important features of biological systems.51 The halogen bond is a unique non-covalent interaction which, although documented for the first time 100 years ago,72 has only flourished during the last two decades.1–10 The investigation of halogen bonding by NMR spectroscopy in solution implies adding an XB donor to a solution of XB acceptor and observation of the behaviour of the chemical shift of the carbon atom to which the halogen atom is attached. The formation of a halogen bond could lead to an increase in the chemical shift of this carbon atom. We considered 13C NMR spectroscopy to be the most accessible and versatile method to probe the XB interaction in solution. Thus, the behaviour of the compounds was investigated by 13C NMR spectroscopy in the presence of selected Lewis bases in CDCl3 or neat deuterated solvents acting as XB acceptors. For example, compound 3 was mixed in CDCl3 with Et3N or 1,4-diazabicyclo[2.2.2]octane (DABCO), or dissolved in pyridine-d5 and dimethylsulfoxide-d6 (DMSO). Fig. 11 presents the 13C NMR spectra of 3 recorded at ambient temperature; they show the shifting of the signal for the C–I carbon atom with a maximum of 6 ppm in the case of pyridine-d5. Compounds 1 and 2 display analogous behaviour observed also in the case of iodinated sydnones.73
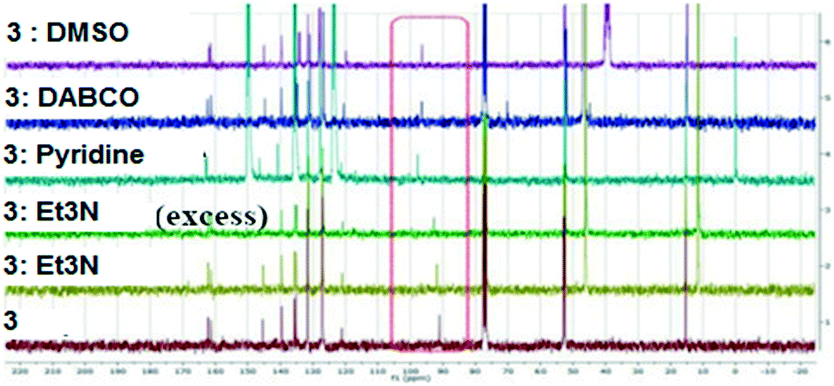 | ||
| Fig. 11 13C NMR spectra of 3 in presence of different XB acceptors as deuterated solvents or dissolved in CDCl3. | ||
The chemical shifts of all the carbon atoms are presented in Table S6 (ESI†) and they unequivocally confirm that only the C-5 carbon atom is influenced by the presence of the base, leading to the conclusion that a strong C–I⋯N (in the case of the amines) or C–I⋯O (in the case of DMSO) interaction have occurred in solution.
We have also investigated the behaviour of 3 upon addition of DABCO in benzene-d6 which was chosen as an inert solvent. This resulted in the deshielding of the C–I carbon atom from 91.6 to 98.6 ppm revealing a deshielding of atom C-5 by 7 ppm. The addition was stopped when no further significant influence on the chemical shift was observed. The addition of DABCO presents also some influences on the 1H NMR spectra which could represent solvent interactions with the H atoms (Fig. 12).
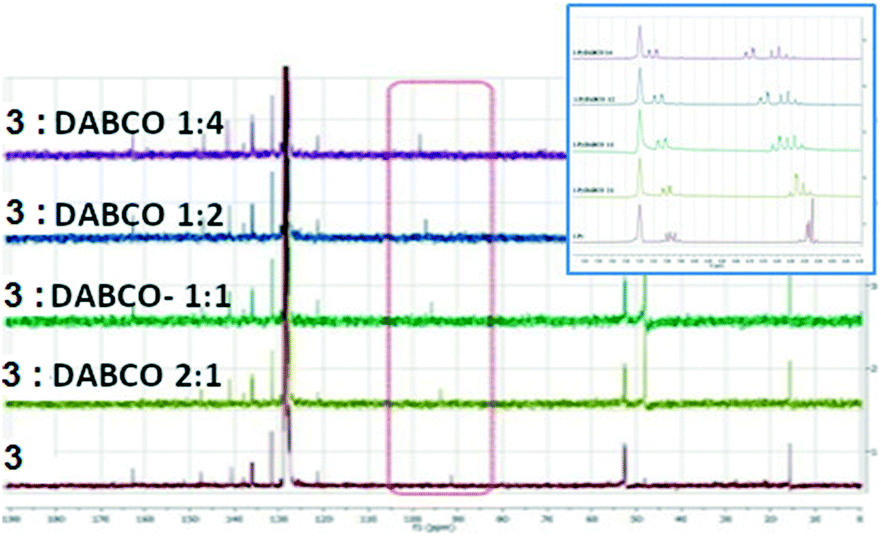 | ||
| Fig. 12 13C NMR spectra of 3 upon the addition of DABCO in C6D6 (inset: the 1H NMR aromatic area). | ||
The magnitude of the chemical shift upon the addition of DABCO assumes the formation of a halogen bonded complex between 3 and DABCO, as proposed in Fig. 13.
 | ||
| Fig. 13 The proposed 1:1 complex of 3 with DABCO in the solution phase. | ||
Table 5 presents the chemical shifts of the carbon C-5 in 3 upon the addition of the different bases or by increasing amount of DABCO in an inert solvent such as C6H6:
| Lewis Base | No base | Et3N | Pyridine-d5 | DMSO | DABCO |
|---|---|---|---|---|---|
| δ C-5 (ppm) | 91.09 | 92.77 | 97.75 | 96.39 | 96.46 |
|
3:DABCO |
No base | 2:1 |
1:1 |
1:2 |
1:4 |
| δ C-5 (ppm) | 91.58 | 93.73 | 95.81 | 97.19 | 98.6 |
In the Tables S6 and S7 (ESI† are presented the chemical shifts of all the atoms in the 1-arylpyrazole 3 in order to confirm that no other atom besides the C-5 is affected by the interaction with the Lewis base.
Conclusions
In conclusion, three highly substituted 5-iodinated 1-arylpyrazoles were investigated for their susceptibility to form halogen bonds. The 5-iodinated compounds present three types of halogen bonding, namely C–I⋯N, C–I⋯O and C–I⋯π respectively. Of course, the solution-state experiments cannot explain the main causes of this variation but certainly in the case of 1-phenylpyrazoles substituted with an iodine atom in position 5, 13C NMR spectroscopy in solution could predict the existence of halogen bonding and also suggest strong interactions which could direct the growth of crystal assemblies. The most important influences seem to be the presence of large substituents at the ortho position of the phenyl ring or the presence of other substituents, such as hydrogen bond donors or acceptors, which may induce competitive interactions. Our investigation, strengthened by a thorough CCDC search and provision of comparative literature data encourage us to further probe the propensity of halogen bond formation in highly substituted iodinated pyrazoles and its importance as a directional force in crystal packing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
MRC is grateful to the NRF (Pretoria) and the University of Cape Town for research support.Notes and references
- G. Cavallo, P. Metrangolo, R. Milani, T. Pilati, A. Priimagi, G. Resnati and G. Terraneo, Chem. Rev., 2016, 116, 2478–2601 CrossRef CAS PubMed.
- G. R. Desiraju, P. S. Ho, L. Kloo, A. C. Legon, R. Marquardt, P. Metrangolo, P. Politzer, G. Resnati and K. Rissanen, Pure Appl. Chem., 2013, 85, 1711–1713 CAS.
- A. Mukherejee, S. Tothadi and G. R. Desiraju, Acc. Chem. Res., 2014, 47, 2514–2524 CrossRef PubMed.
- P. Metrangolo and G. Resnati, IUCrJ, 2014, 1, 5–7 CrossRef CAS PubMed.
- S. J. Grabowski, Phys. Chem. Chem. Phys., 2013, 15, 7249–7259 RSC.
- M. Erdelyi, Chem. Soc. Rev., 2012, 41, 3547–3557 RSC.
- T. M. Beale, M. G. Chudzinski, M. G. Sarwar and M. S. Taylor, Chem. Soc. Rev., 2013, 42, 1667–1680 RSC.
- A.-C. C. Carlsson, A. X. Veiga and M. Erdelyi, Top. Curr. Chem., 2015, 359, 49–76 CrossRef CAS PubMed.
- Y. Lu, H. Li, X. Zhu, W. Zhu and H. Liu, J. Phys. Chem. A, 2011, 115, 4467–4475 CrossRef CAS PubMed.
- T. M. Beale, M. G. Chudzinski, M. G. Sarwar and M. S. Taylor, Chem. Soc. Rev., 2013, 42, 1667–1680 RSC.
- P. Metrangolo, H. Neurkirch, T. Pilati and G. Resnati, Acc. Chem. Res., 2005, 38, 386–395 CrossRef CAS PubMed.
- S. Sirimulla, J. B. Bailey, R. Vegesna and M. Narayan, J. Chem. Inf. Model., 2013, 53, 2781–2791 CrossRef CAS PubMed.
- T. Clark, M. Henneman, J. S. Murray and P. Politzer, J. Mol. Model., 2007, 13, 291–296 CrossRef CAS PubMed.
- A. V. Jentzsch, Pure Appl. Chem., 2015, 87, 15–41 Search PubMed.
- M. J. Langton, S. W. Robinson, M. Igor, V. Felix and P. D. Beer, Nat. Chem., 2014, 6, 1039–1043 CrossRef CAS PubMed.
- M. J. Langton, Y. Xiong and P. D. Beer, Chem. – Eur. J., 2015, 21, 18910–18914 CrossRef CAS PubMed.
- H. Li, Y. Lu, Y. Liu, X. Zhu, H. Liu and W. Zhu, Phys. Chem. Chem. Phys., 2012, 14, 9948–9955 RSC.
- J. F. Bertran and M. Rodriguez, Org. Magn. Reson., 1980, 14, 244–246 CrossRef.
- M. T. Messina, P. Metrangolo, W. Panzeri, E. Ragg and G. Resnati, Tetrahedron Lett., 1998, 39, 9069–9072 CrossRef CAS.
- R. Glasser, N. Chen, H. Wu, N. Knotts and M. Kaupp, J. Am. Chem. Soc., 2004, 126, 4412–4419 CrossRef PubMed.
- J. A. Webb, J. E. Klijn, P. A. Hill, J. L. Bennet and N. S. Goroff, J. Org. Chem., 2004, 69, 660–664 CrossRef CAS PubMed.
- W. N. Moss and N. S. Goroff, J. Org. Chem., 2005, 70, 802–808 CrossRef CAS PubMed.
- I. Castellote, M. Moron, C. Burgos, J. Alvarez-Builla, A. Martin, P. Gomez-Sal and J. J. Vaquero, Chem. Commun., 2007, 1281–1283 RSC.
- A.-C. C. Carlsson, J. Grafenstein, A. Budnjo, J. L. Laurila, J. Bergquist, A. Karim, R. Kleinmaier, U. Brath and M. Erdelyi, J. Am. Chem. Soc., 2012, 134, 5706–5715 CrossRef CAS PubMed.
- Y. Zhang, B. Ji, A. Tian and W. Wang, J. Chem. Phys., 2012, 136, 141101 CrossRef PubMed.
- G. M. Sarwar, D. Ajami, G. Theodorakopoulos, I. D. Petsalakis and J. Rebek Jr., J. Am. Chem. Soc., 2013, 135, 13672–13675 CrossRef PubMed.
- B. Hawthorne, H. Fan-Hagenstein, E. Wood, J. Smith and T. Hanks, Int. J. Spectrosc., 2013, 216518, DOI:10.1155/2013/216518.
- D. A. Smith, L. Brammer, C. A. Hunter and R. N. Perutz, J. Am. Chem. Soc., 2014, 136, 1288–1291 CrossRef CAS PubMed.
- O. Dumele, D. Wu, N. Trapp, N. Goroff and F. Diederich, Org. Lett., 2014, 16, 4722–4725 CrossRef CAS PubMed.
- S. Groni, T. Maby-Raud, C. Fave, M. Branca and B. Schollhorn, Chem. Commun., 2014, 50, 14616–14619 RSC.
- M. Bedin, A. Karim, M. Reitti, A.-C. C. Carlsson, F. Topic, M. Cetina, F. Pan, V. Havel, F. Al-Ameri, V. Sindelar, K. Rissanen, J. Grafenstein and M. Erdelyi, Chem. Sci., 2015, 6, 3746–3756 RSC.
- T.-R. Tero, K. Salorinne, S. Malola and H. Hakkinen, CrystEngComm, 2015, 17, 8231–8241 RSC.
- S. H. Jungbauer, S. Schindler, E. Herdtweck, S. Keller and S. M. Huber, Chem. – Eur. J., 2015, 21, 13625–13636 CrossRef CAS PubMed.
- R. A. Thorson, G. R. Woller, Z. L. Driscoll, B. E. Geiger, C. A. Moss, A. L. Schlapper, E. D. Speetzen, E. Bosch, M. Erdelyi and N. P. Bowling, Eur. J. Org. Chem., 2015, 1685–1695 CrossRef CAS.
- L. Maugeri, J. Asencio-Hernandez, T. Lebl, D. B. Cordes, A. M. Z. Slawin, M.-A. Delsuc and D. Philp, Chem. Sci., 2016, 7, 6422–6428 RSC.
- R. Puttereddy, O. Jurcek, S. Bhowmik, T. Makela and K. Rissanen, Chem. Commun., 2016, 52, 2338–2341 RSC.
- T. L. Ellington, P. L. Reves, B. L. Simms, J. L. Wilson, D. L. Watkins, G. S. Tschumper and N. I. Hammer, ChemPhysChem, 2017, 18, 1267–1273 CrossRef CAS PubMed.
- C. C. Robertson, J. S. Wright, E. J. Carrington, R. N. Perutz, C. A. Hunter and L. Brammer, Chem. Sci., 2017, 8, 5392–5398 RSC.
- M. Kaasik, S. Kaabel, K. Kriis, I. Jarving, R. Aav, K. Rissanen and T. Kanger, Chem. – Eur. J., 2017, 23, 7337–7344 CrossRef CAS PubMed.
- H. Sun, A. Horatscheck, V. Martos, M. Bartetzko, U. Uhrig, D. Lentz, P. Schmeider and M. Nazare, Angew. Chem., Int. Ed., 2017, 56, 6454–6458 CrossRef CAS PubMed.
- P. M. J. Szell, B. Gabidullin and D. Bryce, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2017, 73(Pt 2), 153–162 CrossRef CAS PubMed.
- N. P. Bowling, D. L. Widner, E. R. Robinson, A. B. Perez, H. G. Vang, R. A. Thorson, Z. L. Driscoll, S. M. Giebel, C. W. Berndt, E. Bosch and E. D. Speetzen, Eur. J. Org. Chem., 2017, 5739–5749 Search PubMed.
- S. B. Hakkert, J. Grafenstein and M. Erdelyi, Faraday Discuss., 2017, 203, 333–346 RSC.
- H. Wang, Q. J. Shen and W. Wang, J. Solution Chem., 2017, 46, 1092–1103 CrossRef CAS.
- C. B. Aakeroy, E. P. Hurley and J. Desper, Cryst. Growth Des., 2012, 12, 5806–5814 CrossRef CAS.
- I. Khan and J. M. White, Crystals, 2012, 2, 967–973 CrossRef CAS.
- D. Chand and J. M. Shreeve, Chem. Commun., 2015, 51, 3438–3441 RSC.
- S. A. Surmann and G. Mezei, Acta Crystallogr., Sect. E: Crystallogr. Commun., 2016, 72, 1517–1520 CrossRef CAS PubMed.
- F. Chevallier, Y. S. Halauko, C. Pecceu, I. Nassar, T. U. Dam, T. Roisnel, V. E. Matulis, O. A. Ivashkevich and F. Mongin, Org. Biomol. Chem., 2011, 9, 4671–4684 RSC.
- A. Ansari, M. Asif and Shamsuzzaman, New J. Chem., 2017, 41, 16–41 RSC.
- J. Cerny and P. Hobza, Phys. Chem. Chem. Phys., 2007, 9, 5291–5303 RSC.
- F. Dumitrascu, C. Draghici, D. Dumitrescu, L. Tarko and D. Raileanu, Liebigs Ann./Recl., 1997, 2613–2616 CrossRef CAS.
- (a) F. Dumitrascu, C. I. Mitan, D. Dumitrescu, L. Barbu, M. Hrubaru and D. Caproiu, Rev. Chim., 2003, 54, 747 CAS; (b) F. Dumitrascu, C. Draghici, C. Crangus, M. T. Caproiu, C. I. Mitan, D. Dumitrescu and D. Raileanu, Rev. Roum. Chim., 2002, 47, 315–318 Search PubMed; (c) D. L. Browne and J. P. A. Harrity, Tetrahedron, 2010, 66, 533–568 CrossRef.
- A. Bondi, J. Phys. Chem, 1964, 68, 441–451 CrossRef CAS.
- (a) S. K. Wolff, D. J. Grimwood, J. J. McKinnon, M. J. Turner, D. Jayatilaka and M. A. Spackman, CrystalExplorer (Version 3.1), University of Western, Australia, 2012 Search PubMed; (b) M. A. Spackman and D. Jayatilaka, CrystEngComm, 2009, 11, 19–32 RSC.
- (a) J. J. McKinnon, M. A. Spackman and A. S. Mitchell, Acta Crystallogr., Sect. B: Struct. Sci., 2004, 60, 627–668 CrossRef PubMed; (b) M. A. Spackman and J. J. McKinnon, CrystEngComm, 2002, 4, 378–392 RSC.
- (a) H. Y. Gao, X. R. Zhao, H. Wang, X. Pang and W. J. Jin, Cryst. Growth Des., 2012, 12, 4377–4387 CrossRef CAS; (b) Q. J. Shen, X. Pang, X. R. Zhao, H. Y. Gao, H.-L. Sun and W. J. Jin, CrystEngComm, 2012, 14, 5027–5034 RSC.
- F. H. Allen, C. A. Baalham, J. P. M. Lommerse and P. R. Raithby, Acta Crystallogr., Sect. B: Struct. Sci., 1998, 54, 320–329 CrossRef.
- M. Egli and S. Sarkhel, Acc. Chem. Res., 2007, 40, 197–205 CrossRef CAS PubMed.
- J. S. Murray, L. Macaveiu and P. Politzer, J. Comput. Sci., 2014, 5, 590–596 CrossRef.
- M. H. Kolar and P. Hobza, Chem. Rev., 2016, 116, 5155–5187 CrossRef CAS PubMed.
- P. Politzer, J. S. Murray, T. Clark and G. Resnati, Phys. Chem. Chem. Phys., 2017, 19, 32166–32178 RSC.
- M. J. Frisch et al., Gaussian 09, Revision C.01, Wallingford, CT, 2009 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B, 1988, 37, 785–789 CrossRef CAS PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- N. Godbout, D. R. Salahub, J. Andzelm and E. Wimmer, Can. J. Chem., 1992, 70, 560–571 CrossRef CAS.
- R. F. W. Bader, M. T. Carroll, J. R. Cheeesman and C. Chang, J. Am. Chem. Soc., 1987, 109, 7968–7979 CrossRef CAS.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- C. I. Nwachukwu, N. P. Bowling and E. Bosch, Acta Crystallogr., Sect. C: Struct. Chem., 2017, 73, 2–8 CrossRef CAS PubMed.
- S. F. Boys and F. Bernardi, Mol. Phys., 1970, 19, 553–566 CrossRef CAS.
- M. M. Colin and H. Gaultier de Claubry, Ann. Chim., 1814, 90, 87–100 Search PubMed.
- C. Draghici, M. R. Caira, D. E. Dumitrescu and F. Dumitrascu, Rev. Chim., 2018, 69, 843–846 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: CCDC 1939541–1939543. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9ce01263j |
| This journal is © The Royal Society of Chemistry 2019 |