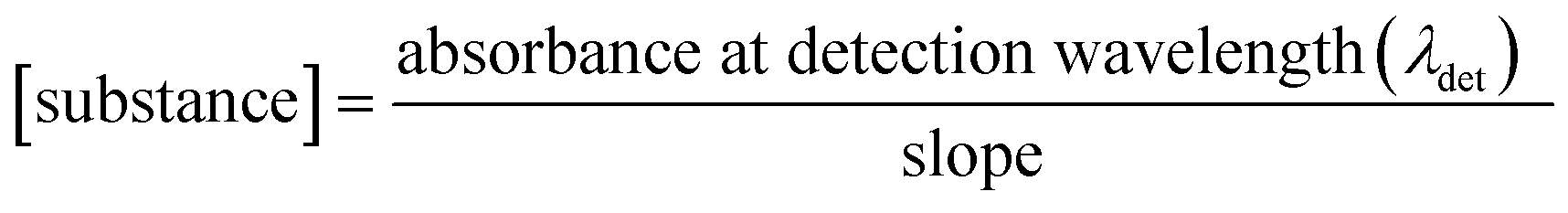DOI:
10.1039/C9CE01195A
(Paper)
CrystEngComm, 2019,
21, 7373-7388
Synthon hierarchy in theobromine cocrystals with hydroxybenzoic acids as coformers†
Received
31st July 2019
, Accepted 9th October 2019
First published on 20th November 2019
Abstract
Pharmaceutical cocrystals, multicomponent solids composed of molecular and/or ionic compounds connected by noncovalent interactions, are objects of interest in crystal engineering. Theobromine, as an active pharmaceutical ingredient, was used for cocrystallization with dihydroxybenzoic acids as crystal coformers. All of these dimethylxanthine derivatives were obtained by slow evaporation from solution and they were structurally-characterized by a single X-ray diffraction method. Solid-state synthesis products through grinding (green chemistry experiments) were confirmed by powder X-ray diffraction measurements. Simultaneous thermal analyses for samples from grinding were performed. The various supramolecular synthons formed by theobromine responsible for the arrangement of molecules in the crystal lattice of its cocrystals were specified. The hydrogen-bonded motifs present in these cocrystals together with theobromine–acid solids deposited in the CSD were summarized. Furthermore, UV-vis spectra measurements were made to check the change in the solubility of theobromine after its cocrystallization.
1. Introduction
The production of drugs is extremely important nowadays. Chemical stability, high solubility and, hence, high bioavailability are some of the most important properties that should be characteristic of a particular active pharmaceutical ingredient (API).1 Amidon et al. introduced the classification of APIs into 4 groups due to the above parameters: class 1 (high solubility, high permeability), class 2 (low solubility, high permeability), class 3 (high solubility, low permeability) and class 4 (low solubility, low permeability).2 About 40% of sold medicines and 80–90% of drugs in the production process are poorly soluble in water.3 A major problem for the pharmaceutical industry is the lack of desired physicochemical properties for given APIs, because it requires searching for suitable methods to improve them,4i.e. obtaining polymorphic forms,5–13 solvates and hydrates,14–21 cocrystals22–26 or salts.27–30
Polymorphs of APIs have different properties, such as hygroscopicity, stability, solubility or processability, but, in many cases, it is not possible to predict or design a specific polymorphic form of an API. Moreover, some of them may be metastable and may undergo phase transformations,31,32 which may result in undesirable changes in physicochemical properties.33 Solvent molecules in solvates can improve the stability of metastable forms through strong hydrogen bonds with API molecules, or they can act as guests by filling free places in the host crystal lattice without interactions between them.34,35 However, solvents can sometimes lead to disorder in the crystal lattice, which can result in a metastable form of a substance.36 In contrast to cocrystals, preparation of salts requires APIs having ionizable functional groups.4 Therefore, the cocrystallization of drugs offers more synthetic possibilities. Another advantage is that a particular API can exist in a stable crystalline form with a suitably selected coformer without excipients.37 What is more, it is a relatively fast and simple method, which in turn translates into financial issues for pharmaceutical companies and can improve properties such as tabletability, stability, solubility, dissolution rates, bioavailability and mechanical properties.38–41
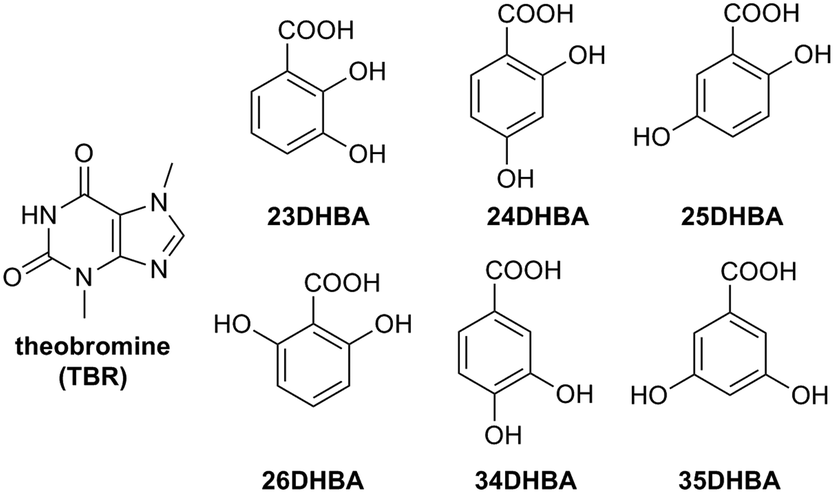
In this paper, four theobromine (TBR) cocrystals with 2,4-dihydroxy- (24DHBA), 2,5-dihydroxy- (25DHBA), 3,4-dihydroxy- (34DHBA) and 3,5-dihydroxybenzoic acid (35DHBA), a cocrystal hydrate with 2,3-dihydroxybenzoic acid (23DHBA), and a salt monohydrate containing 2,6-dihydroxybenzoate anion (26DHBA) were reported. All of these xanthine derivatives obtained by cocrystallization from solution and by grinding were characterized by SXRD, PXRD and UV-vis spectral measurements to check the improvement of their solubility in water after cocrystallization. In earlier work, we report theobromine with monohydroxybenzoic acid cocrystals.42 Both of these studies fit into the mainstream of structural studies of purine alkaloid cocrystals.29,43,44 Their aim is to hierarchize supramolecular synthons responsible for the molecular arrangement in xanthine cocrystals. The design of new pharmaceutical derivatives with the desired physicochemical properties, having two or more components, could be simpler with a greater understanding and knowledge of the synthonic hierarchy in organic cocrystals.45–47
The choice of theobromine for cocrystallization is related to the fact that only 8 cocrystals (with 5-chlorosalicylic,48 2-hydroxybenzoic,49 oxalic,50 trifluoroacetic,51 malonic,51 acetic,52 and anthranilic49,53 acids and melamine54) and 4 cocrystal hydrates (with quercetin,55 vanillin,56 vanillic acid57 and 3,4,5-trihydroxybenzoic acid55) with this alkaloid have been deposited in the CSD so far. Hydroxybenzoic acids are often used as coformers to obtain multi-component systems, e.g. with theophylline,43 caffeine,44,58 pyrazinamide,59,60 urotropine,61–63 ethenzamide64 and gabapentin.65
2. Experimental section
2.1. Materials
Theobromine (99%) was purchased from Swiss Herbal Institute. 2,3-Dihydroxy- (98%), 2,4-dihydroxy- (95%), 2,5-dihydroxy- (99%), 2,6-dihydroxy- (98%), 3,4-dihydroxy- (95%) and 3,5-dihydroxybenzoic acid (97%) were obtained from TriMen Chemicals and they were used without purification. Methanol and acetonitrile were purchased from Chempur. Millipore distilled water (18 MΩ cm) was used in all absorption experiments.
2.2. Cocrystallization from solution
The stoichiometric ratios of theobromine and a particular dihydroxybenzoic acid were used (Fig. 1). TBR (19.3 mg, 0.107 mmol) with 23DHBA (16.4 mg, 0.106 mmol), TBR (18.7 mg, 0.104 mmol) with 24DHBA (16.2 mg, 0.105 mmol), TBR (19.6 mg, 0.109 mmol) with 25DHBA (17 mg, 0.110 mmol), TBR (18.6 mg, 0.103 mmol) with 34DHBA (16.2 mg, 0.105 mmol), and TBR (19.6 mg, 0.109 mmol) with 35DHBA (17 mg, 0.110 mmol) were dissolved in methanol–water solution, whereas TBR (19.6 mg, 0.109 mmol) with 26 DHBA (16.6 mg, 0.108 mmol) was dissolved in acetonitrile–water solution by heating and stirring. The resulting clear solutions were filtered. Single crystals of (TBR)·(23DHBA)·(H2O), (TBR)·(24DHBA), (TBR)·(25DHBA), (TBR-H)+·(26DHBA)−·(H2O), (TBR)·(34DHBA) and (TBR)·(35DHBA) were obtained by slow evaporation of filtrates under ambient conditions within 3–7 days.
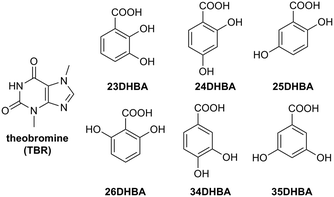 |
| | Fig. 1 Theobromine and dihydroxybenzoic acids. | |
2.3. Cocrystallization by grinding
An oscillatory ball mill (Retsch MM300) was used for milling experiments. The stoichiometric ratios of theobromine and the given dihydroxybenzoic acid were placed in stainless steel milling jars. TBR (15.1 mg, 0.083 mmol) with 23DHBA (12.9 mg, 0.084 mmol) and TBR (19.6 mg, 0.109 mmol) with 26DHBA (16.8 mg, 0.109 mmol) were ground with the addition of 20 μl of water. Neat grinding processes were performed using TBR (14.2 mg, 0.079 mmol) with 24DHBA (12.1 mg, 0.079 mmol), TBR (27.2 mg, 0.151 mmol) with 25DHBA (23.1 mg, 0.150 mmol), TBR (16.6 mg, 0.092 mmol) with 34DHBA (14.3 mg, 0.093 mmol) and TBR (8.6 mg, 0.048 mmol) with 35DHBA (7.5 mg, 0.049 mmol). Each grinding was carried out for 60 minutes at a frequency of 25 Hz using two 4.8 mm stainless steel balls.
2.4. Single crystal X-ray diffraction
Low-temperature measurements were carried out using an Oxford Diffraction SuperNova diffractometer with monochromatic CuKα radiation (1.54184 Å) and a Cryojet cooling system. Data collection and data reduction were performed using CrysAlisPro66 and CrysAlisRed67 programs, respectively. The crystal structures were solved by intrinsic phasing using SHELXT-2015 and were refined using the least-squares method with SHELXL-2015 software.68 Solution, refinement and structural analyses were carried out using the Olex2 program.69 All non-hydrogen atoms were refined with anisotropic displacement parameters. All hydrogen atoms were derived from the difference Fourier map but, finally, in (TBR)·(24DHBA) and (TBR)·(23DHBA)·(H2O), hydrogen atoms bonded to carbon atoms were positioned geometrically using AFIX commands and were refined using a riding-hydrogen model with Uiso(H) = 1.2Ueq(C) for aromatic and imidazole H atoms or Uiso(H) = 1.5Ueq(C) for hydrogen atoms belonging to the methyl group. In these two crystals, hydrogen atoms connected with heteroatoms were refined without constraints. In the rest of the theobromine derivatives, all hydrogen atoms were refined isotropically. Crystallographic data are presented in Table 1.
Table 1 Crystallographic data and refinement details for the described theobromine complexes
|
|
(TBR)·(23DHBA)·(H2O) |
(TBR)·(24DHBA) |
(TBR)·(25DHBA) |
(TBR-H)+·(26DHBA)−·(H2O) |
(TBR)·(34DHBA) |
(TBR)·(35DHBA) |
| Molecular formula |
(C7H8N4O2)·(C7H6O4)·(H2O) |
(C7H8N4O2)·(C7H6O4) |
(C7H8N4O2)·(C7H6O4) |
(C7H9N4O2)·(C7H5O4)·(H2O) |
(C7H8N4O2)·(C7H6O4) |
(C7H8N4O2)·(C7H6O4) |
|
M
r, g mol−1 |
352.31 |
334.29 |
334.29 |
352.31 |
334.29 |
334.29 |
| Crystal system |
Monoclinic |
Monoclinic |
Triclinic |
Monoclinic |
Orthorhombic |
Monoclinic |
| Space group |
Cc
|
P21/n |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P21/n |
Pbcn
|
P21/c |
|
a, Å |
13.2358(3) |
7.9392(3) |
6.8492(3) |
15.2976(3) |
26.7527(5) |
14.4617(3) |
|
b, Å |
15.2794(2) |
6.3569(2) |
7.9605(4) |
6.8450(1) |
14.2120(2) |
12.7978(3) |
|
c, Å |
7.8074(2) |
28.7601(7) |
14.0276(8) |
15.9300(3) |
7.2608(2) |
15.7666(3) |
|
α, ° |
90 |
90 |
79.391(4) |
90 |
90 |
90 |
|
β, ° |
106.180(2) |
96.093(3) |
78.550(4) |
115.911(3) |
90 |
92.3554(17) |
|
γ, ° |
90 |
90 |
77.232(4) |
90 |
90 |
90 |
|
V, Å3 |
1516.39(6) |
1443.29(8) |
723.16(7) |
1500.38(6) |
2760.62(10) |
2915.58(10) |
|
Z, Z' |
4, 1 |
4, 1 |
2, 1 |
4, 1 |
8, 1 |
8, 2 |
|
D
x, g cm−1 |
1.543 |
1.538 |
1.535 |
1.560 |
1.609 |
1.523 |
|
F(000) |
736 |
696 |
348 |
736 |
1392 |
1392 |
|
μ(Cu Kα), mm−1 |
1.08 |
1.05 |
1.05 |
1.09 |
1.10 |
1.04 |
|
T, K |
132(2) |
132(1) |
130.3(4) |
131.8(3) |
130.9(8) |
130.8(6) |
| Crystal size, mm3 |
0.15 × 0.20 × 0.26 |
0.30 × 0.07 × 0.03 |
0.28 × 0.10 × 0.04 |
0.30 × 0.12 × 0.05 |
0.33 × 0.05 × 0.03 |
0.14 × 0.12 × 0.12 |
|
Θ range for data collection, ° |
4.5–75.9 |
3.1–76.1 |
3.3–76.4 |
3.3–76.3 |
3.3–76.4 |
3.1–76.1 |
| Range of indices (h, k, l) |
−16 → 16, −19 → 13, −9 → 9 |
−9 → 9, −7 → 7, −36 → 22 |
−8 → 8, −9 → 9, −17 → 17 |
−19 → 15, −8 → 8, −19 → 17 |
−33 → 29, −14 → 17, −8 → 7 |
−17 → 18, −16 → 14, −18 → 19 |
| Collected reflections |
7869 |
5993 |
11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 318 318 |
6426 |
7196 |
16119 |
| Unique reflections |
2847 |
2932 |
2963 |
3062 |
2827 |
6048 |
| Reflections with I > 2σ(I) |
2834 |
2574 |
2678 |
2766 |
2530 |
5330 |
|
R
int
|
0.017 |
0.018 |
0.027 |
0.019 |
0.037 |
0.023 |
| No. of parameters |
252 |
235 |
273 |
290 |
273 |
545 |
|
R indices with I > 2σ(I) |
R
1 = 0.0260, wR2 = 0.0728 |
R
1 = 0.0464, wR2 = 0.1363 |
R
1 = 0.0519, wR2 = 0.1499 |
R
1 = 0.0366, wR2 = 0.1018 |
R
1 = 0.0533, wR2 = 0.1441 |
R
1 = 0.0470, wR2 = 0.1302 |
|
R indices with all data |
R
1 = 0.0261, wR2 = 0.0730 |
R
1 = 0.0542, wR2 = 0.1414 |
R
1 = 0.0556, wR2 = 0.1553 |
R
1 = 0.0399, wR2 = 0.1051 |
R
1 = 0.0594, wR2 = 0.1478 |
R
1 = 0.0529, wR2 = 0.1354 |
| GOF |
1.064 |
1.095 |
1.047 |
1.075 |
1.09 |
1.08 |
| Δρmin., Δρmax, e Å−3 |
−0.18, 0.20 |
−0.23, 0.53 |
−0.32, 0.41 |
−0.25, 0.25 |
−0.28, 0.25 |
−0.29, 0.32 |
| CCDC deposit no. |
1938121
|
1938122
|
1938144
|
1938146
|
1938148
|
1938149
|
2.5. Powder X-ray diffraction (PXRD)
An Oxford Diffraction Xcalibur diffractometer with a MoKα radiation source (λ = 0.71073 Å) was used for powder diffraction of samples from grinding. Measurements were performed at room temperature. Experimental conditions: scanning intervals 5–40° (2θ), step between thetas 0.01 and time per step 0.5 s. For data collection, CrysAlisPro28 software was used.66 Analysis and comparison of experimental powder XRD patterns from grinding and calculated powder XRD patterns from the crystal structure were made using Kdif software.70 Theoretical patterns were determined using the Mercury program.71
2.6. Solubility studies of cocrystals by steady-state absorption spectroscopy
Steady-state UV-vis spectroscopy was used to determine the cocrystal solubility in distilled water. UV-vis absorption spectra were recorded using a two-beam Cary 100 UV-vis spectrometer scanning from 200 to 800 nm with 1 nm increments. Quartz cells with an optical length of 2 mm were used. Calibration curves of every cocrystal were prepared (Fig. S1†). Substance concentrations versus absorbance of the substance at detection wavelength were plotted (Table 3). A linear relationship was obtained and the slope was calculated from the graph. To determine the solubility of cocrystals, saturated aqueous solutions of each were prepared. The absorbance at detection wavelength (λdet) was measured and the concentration of the substance was calculated by applying the following relationship:| |  | (1) |
2.7. Simultaneous thermal analysis (STA)
The thermal properties of the samples from mechanical grinding were characterized using a STA analyser (Perkin-Elmer STA6000). The thermal measurements were carried out under a nitrogen atmosphere from room temperature to 400 °C at 10 °C min−1.
3. Results
In this paper we report four cocrystals of theobromine (TBR) with 2,4-dihydroxy- (24DHBA), 2,5-dihydroxy- (25DHBA), 3,4-dihydroxy- (34DHBA) and 3,5-dihydroxybenzoic acid (35DHBA), one cocrystal hydrate with 2,3-dihydroxybenzoic acid (23DHBA) and one salt monohydrate containing 2,6-dihydroxybenzoate anion (26DHBA).
The nature of the above complexes has been ambiguously confirmed based on the geometry of the carboxyl group and the carboxylic acid proton location. In (TBR-H)+·(26DHBA)−·H2O the acidic proton was localized on a difference Fourier map near the imidazole nitrogen atom.
An empirical parameter which can help in acid–base system design and gives an indication of the interval values, where proton transfer between an acid and base can be observed, is the ΔpKa parameter73 described by the equation:
| | | ΔpKa = pKa(protonated base) − pKa(acid) | (2) |
All ΔpKa values for our theobromine derivatives indicated that cocrystals would be formed (Table 4). The latest studies about the ΔpKa rule on crystal design have shown that a salt will form when ΔpKa > 4 and a cocrystal when ΔpKa < −1 (based on 6465 acid–base complexes deposited in the CSD).73,74 In Cruz-Cabeza work73 ionizable and neutral complexes as a function of the calculated ΔpKa graph were presented and it can be concluded that salt formation is even possible, when this value is equal to about −3. For 2,6-dihydroxybenzoic acid and theobromine this value is equal to −2.21 and a typical salt was formed. Considering three principles of hydrogen bond formation in organic compounds established by Etter:75
1. “All good proton donors and acceptors are used in hydrogen bonding.”
2. “6-membered-ring intramolecular hydrogen bonds form in preference to intermolecular hydrogen bonds.”
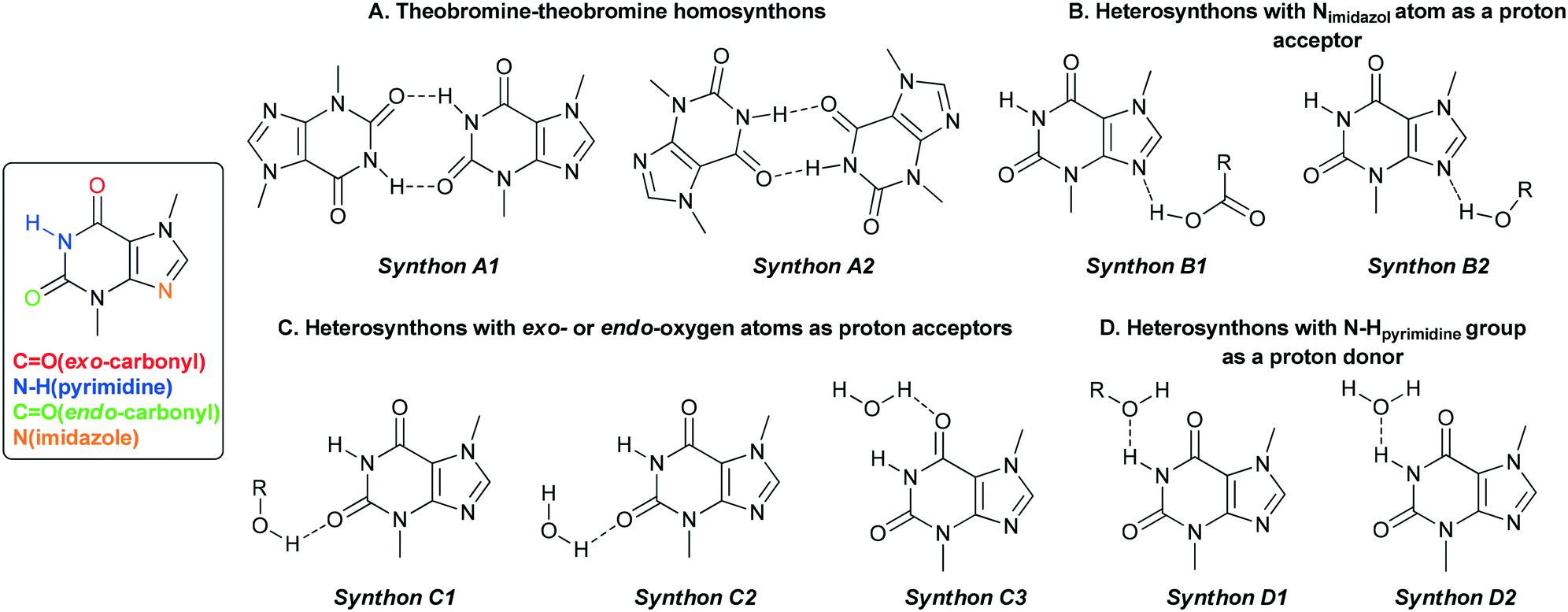
3. “The best proton donors and acceptors remaining after intramolecular hydrogen-bond formation form intermolecular hydrogen bonds to one another.” and the structure of selected coformers, it can be concluded that TBR has one good proton donor (N–H group in the pyrimidine ring) and three good proton acceptors (exo- and endo-carbonyl oxygen atoms, and the imidazole nitrogen atom), while dihydroxybenzoic acids have three good proton donors (hydroxyl groups) and one good proton acceptor (carbonyl oxygen atom in the carboxyl group). Water molecules can also play a very important role in crystal structure formation, because they can be both hydrogen bond acceptors and donors. The occurrence and role of particular synthons in the described cocrystal structures will be discussed below.
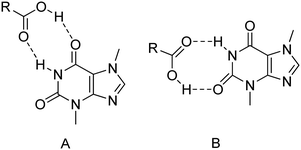
3.1. Structural characterization of theobromine cocrystals
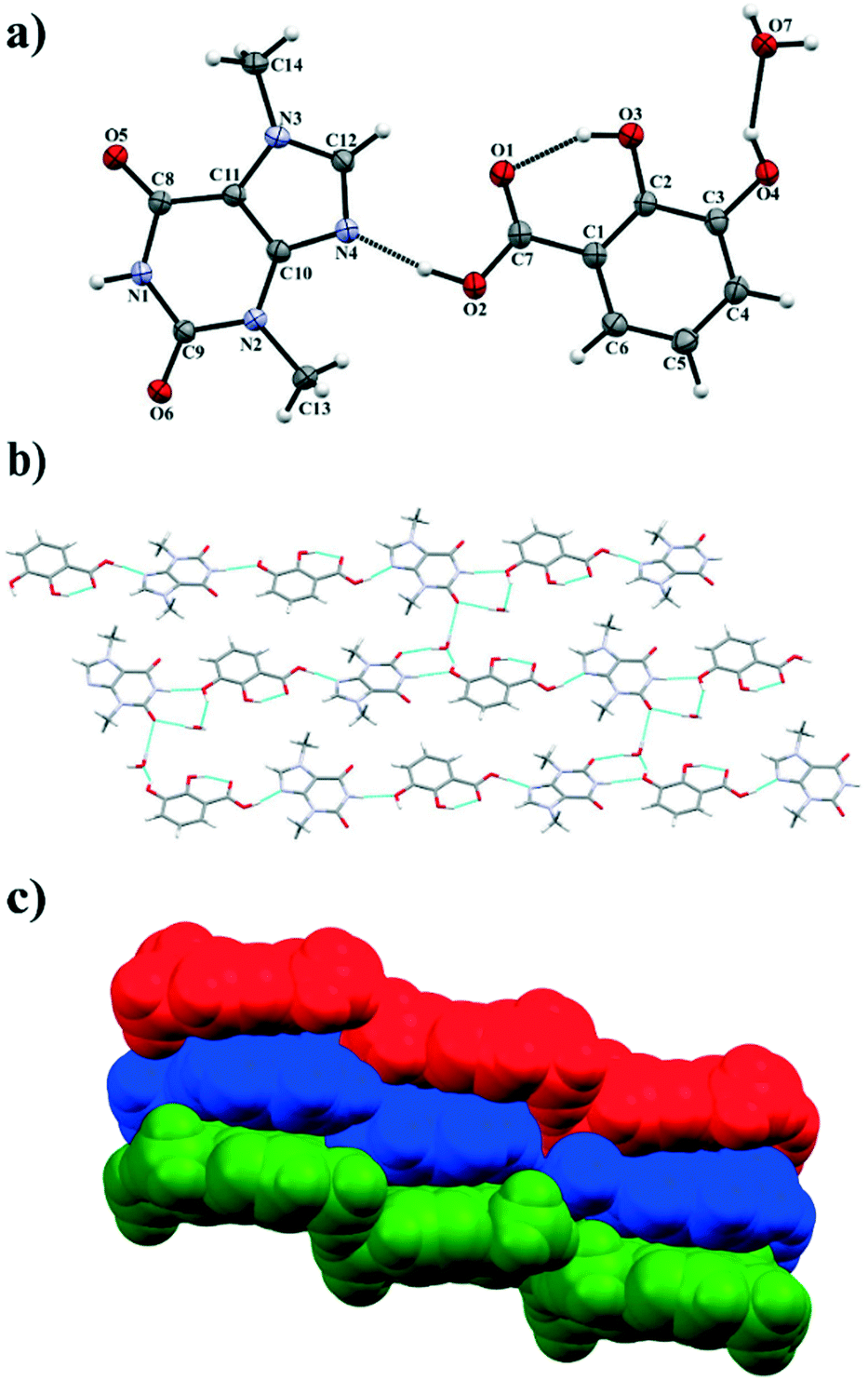
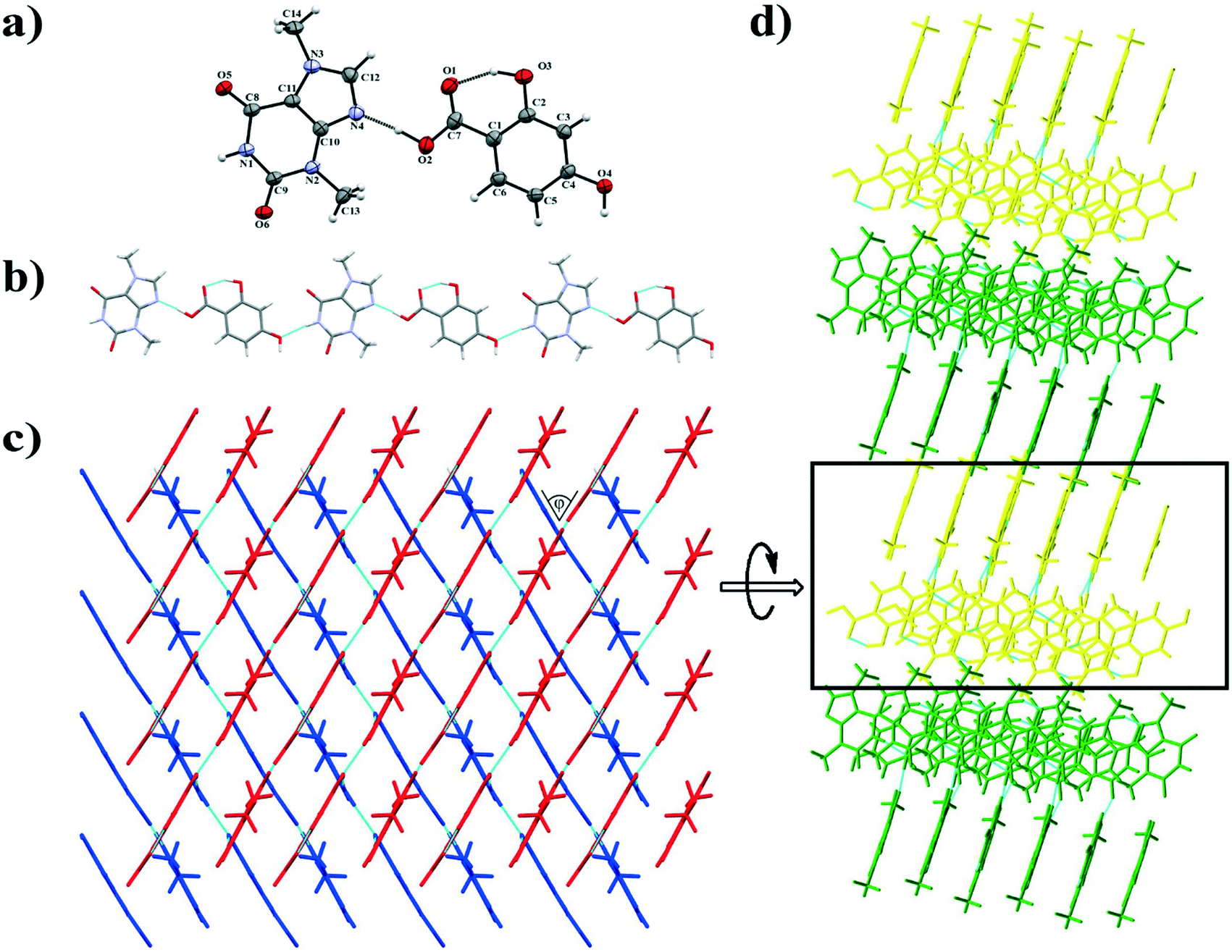
3.1.1. Theobromine-2,3-dihydroxybenzoic acid monohydrate.
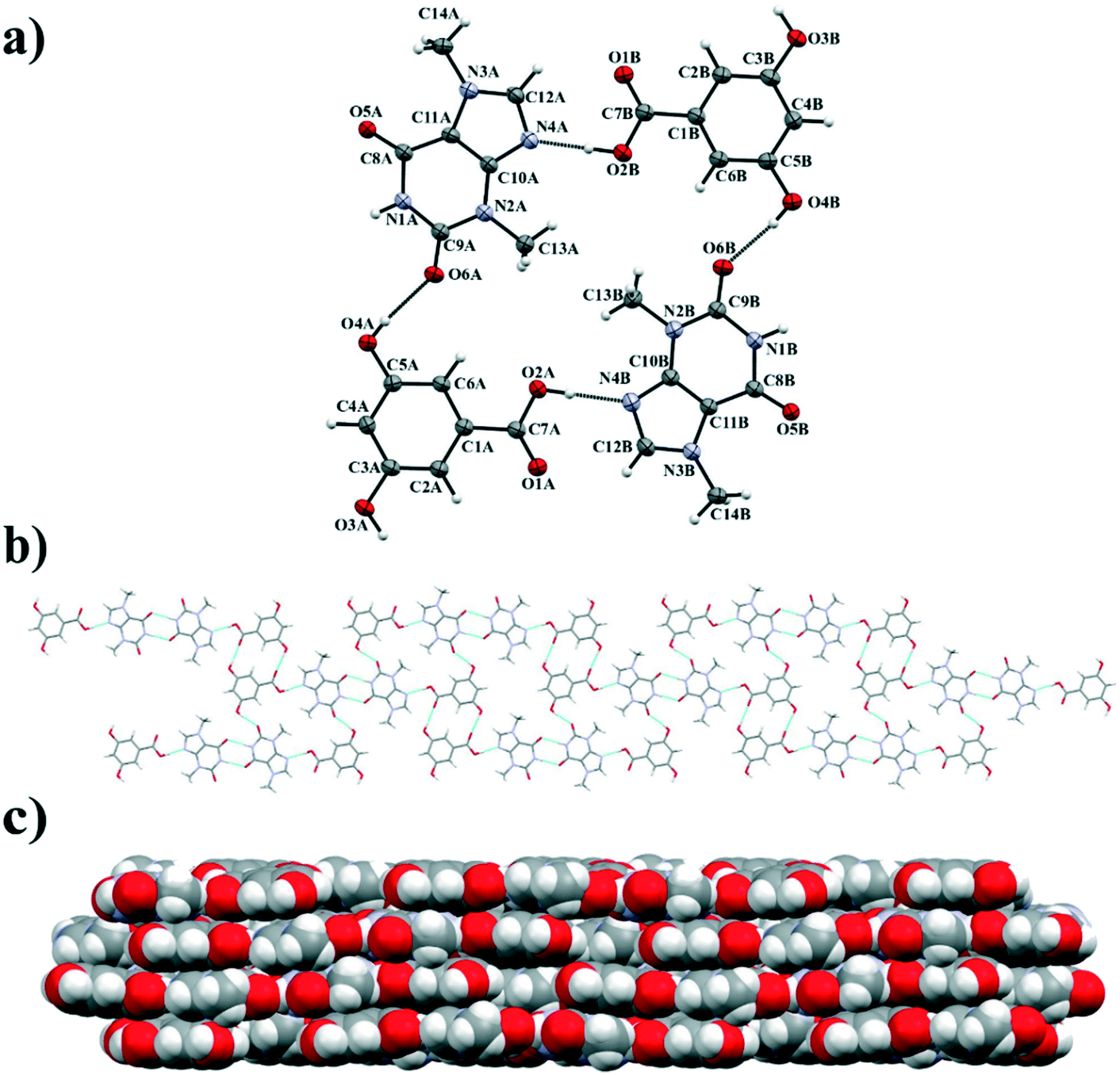
TBR and 23DHBA cocrystallize as a monohydrate in the monoclinic, noncentrosymmetric Cc space group in a 1:1:1 stoichiometric ratio (Fig. 3a). The components form a 1D polymer chain (Fig. 3b). The TBR molecule is hydrogen bonded via O2–H2⋯N4 interaction with the 23DHBA molecule (synthon B1 – Fig. 2, Table 2). The N–H group in the alkaloid pyrimidine ring is the proton donor to the oxygen atom of the meta-hydroxyl group in the acid (N1–H1⋯O4i hydrogen bond, synthon D1). This hydroxyl group is connected with a water molecule by the O4–H4⋯O7 hydrogen bond. At the same time the solvent molecule is the proton donor to the oxygen atom of the endo-carbonyl group in the TBR molecule (O7–H7A⋯O6ii interaction, synthon C2). In this way an R33(8) cyclic array is formed. Additionally, the intramolecular hydrogen bond O3–H3⋯O1 in the 23DHBA molecule leads to S11(6) ring formation. The water molecule is also the donor of the second proton to the oxygen atom of the endo-carbonyl group of TBR located in the neighboring chain (O7–H7A⋯O6 hydrogen bond, synthon C2). So, the DDA (donor–donor–acceptor) hydrogen-bonding character of solvent molecules can be observed and it is responsible for the 2D structure formation. 1D polymer systems are arranged in a “stair” motif, and by π(TBR)⋯π(23DHBA) interactions a 3D network is formed (Fig. 3c, Table S1†).
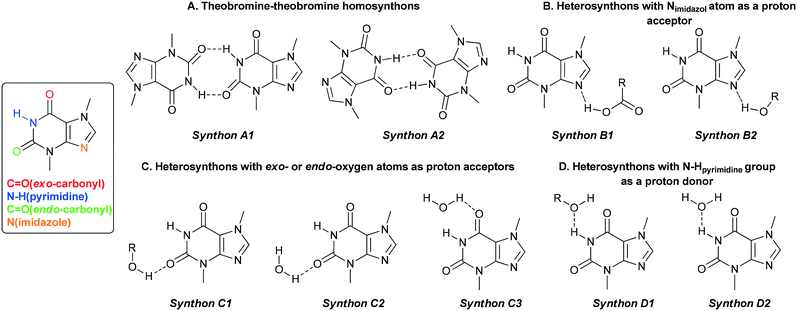 |
| | Fig. 2 Particular theobromine synthons identified in its cocrystals with mono-42 and dihydroxybenzoic acids. | |
Table 2 Hydrogen bond parameters in the described theobromine cocrystals
| Cocrystal |
D–H⋯A |
D–H [Å] |
H⋯A [Å] |
D⋯A [Å] |
D–H⋯A [°] |
| TBR·23DHBA·H2O |
N1–H1⋯O4i |
0.87(3) |
2.04(3) |
2.898(2) |
172(3) |
| O7–H7A⋯O6ii |
0.88(4) |
1.99(4) |
2.857(2) |
168(3) |
| O2–H2⋯N4 |
1.03(6) |
1.63(6) |
2.657(3) |
174(5) |
| O7–H7B⋯O6iii |
0.81(4) |
2.06(4) |
2.867(2) |
177(4) |
| O4–H4⋯O7 |
0.87(3) |
1.89(3) |
2.689(2) |
152(3) |
| O3–H3⋯O1 |
0.87(5) |
1.76(5) |
2.566(2) |
153(4) |
| Symmetry codes: (i) x − 1/2, −y + 1/2, z + 3/2; (ii) x + 1/2, −y + 1/2, z − 3/2; (iii) x + 1/2, y − 1/2, z − 1 |
| TBR·24DHBA |
N1–H1⋯O4i |
0.89(3) |
2.02(3) |
2.899(2) |
172(2) |
| O4–H4⋯O6ii |
0.91(3) |
1.77(3) |
2.673(2) |
174(3) |
| O3–H3⋯O1 |
1.04(2) |
1.63(3) |
2.584(2) |
150(3) |
| O2–H2⋯N4 |
1.06(4) |
1.59(4) |
2.648(2) |
173(3) |
| Symmetry codes: (i) x + 1, y + 2, z; (ii) −x + 1/2, y − 3/2, −z + 1/2 |
| TBR·25DHBA |
O4–H4⋯O6i |
0.85(3) |
1.91(3) |
2.748(2) |
168(2) |
| N1–H1⋯O5ii |
0.87(3) |
1.92(3) |
2.781(2) |
172(2) |
| O3–H3⋯O1 |
0.94(3) |
1.75(3) |
2.602(2) |
150(3) |
| O2–H2⋯N4 |
0.96(3) |
1.68(3) |
2.628(2) |
171(3) |
| Symmetry codes: (i) −x + 1, −y + 2, −z + 1; (ii) −x + 2, −y + 2, −z |
| (TBR-H)+·(26DHBA)−·H2O |
N1–H1⋯O7 |
0.92(2) |
1.81(2) |
2.735(2) |
179(2) |
| O7–H7A⋯O4i |
0.93(2) |
1.90(2) |
2.826(2) |
177(2) |
| O7–H7B⋯O5ii |
0.86(3) |
1.98(3) |
2.827(2) |
167(3) |
| O4–H4A⋯O2 |
0.95(3) |
1.68(3) |
2.549(1) |
150(2) |
| O3–H3⋯O1 |
0.91(2) |
1.68(2) |
2.554(2) |
161(2) |
| N4–H4⋯O2 |
1.06(3) |
1.50(3) |
2.564(2) |
180(2) |
| Symmetry codes: (i) −x + 1/2, y − 1/2, −z + 1/2; (ii) −x, −y + 1, −z + 1 |
| TBR·34DHBA |
O4–H4⋯O1i |
0.84(3) |
1.91(4) |
2.731(2) |
164(3) |
| O3–H3⋯O6ii |
0.95(4) |
1.83(4) |
2.783(2) |
178(3) |
| N1–H1⋯O5iii |
0.85(3) |
1.89(4) |
2.742(2) |
175(3) |
| O2–H2⋯N4 |
0.90(4) |
1.77(4) |
2.670(2) |
178(4) |
| Symmetry codes: (i) −x + 1/2, y + 1/2, z; (ii) −x + 1, −y + 1, −z + 1; (iii) −x + 1, −y, −z + 1 |
| TBR·35DHBA |
O2A–H2A⋯N4B |
0.91(3) |
1.78(3) |
2.690(2) |
178(2) |
| O3A–H3A⋯O1Bi |
0.95(4) |
1.87(4) |
2.795(2) |
164(3) |
| O4A–H4A⋯O6A |
0.81(3) |
1.96(3) |
2.757(2) |
172(3) |
| O2B–H2B⋯N4A |
0.98(3) |
1.71(3) |
2.689(2) |
178(3) |
| O3B–H3B⋯O1Aii |
0.95(4) |
1.85(4) |
2.789(2) |
173(3) |
| O4B–H4B⋯O6B |
0.83(3) |
1.92(3) |
2.737(2) |
170(3) |
| N1A–H1A⋯O5Biii |
0.87(2) |
1.92(2) |
2.785(2) |
176(2) |
| N1B–H1B⋯O5Aiv |
0.89(3) |
1.93(3) |
2.811(2) |
176(2) |
| Symmetry codes: (i) x, y, z + 1; (ii) x, y, z − 1; (iii) x − 1, y, z; (iv) x + 1, y, z |
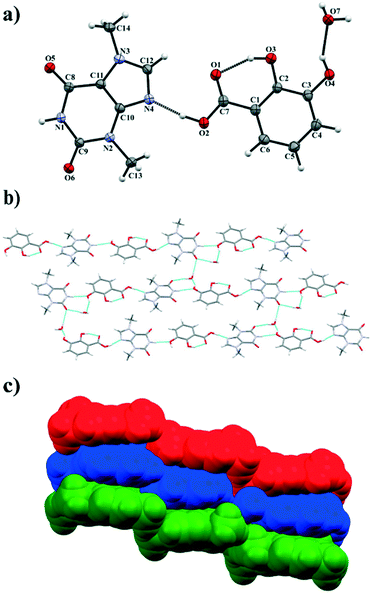 |
| | Fig. 3 a) ORTEP representation showing the TBR·23DHBA·H2O asymmetric unit with an atomic numbering scheme (thermal ellipsoids are plotted with the 50% probability level); b) two-dimensional structure showing a “stair” motif composed of 1D polymer systems connected by water molecules; c) 3D structure of TBR·23DHBA·H2O cocrystal hydrate held by π-stacking interactions. | |
Table 3 Cocrystal detection wavelengths
|
|
Detection wavelength (λdet) |
| TBR·23DHBA·H2O |
340 |
| TBR·24DHBA |
315 |
| TBR·25DHBA |
323 |
| (TBR-H)+·(26DHBA)−·H2O |
340 |
| TBR·34DHBA |
310 |
| TBR·35DHBA |
323 |
Table 4 Calculated ΔpKa values
| Acid |
pKaacida |
Calculated ΔpKab |
|
pKa values for dihydroxybenzoic acids were taken from D.-K. Bučar et al. publication.43
pKa (protonated base) for theobromine is equal to −0.91.72
|
| 23DHBA |
2.96 |
−3.87 |
| 24DHBA |
3.32 |
−4.23 |
| 25DHBA |
3.01 |
−3.92 |
| 26DHBA |
1.30 |
−2.21 |
| 34DHBA |
4.45 |
−5.36 |
| 35DHBA |
3.96 |
−4.87 |
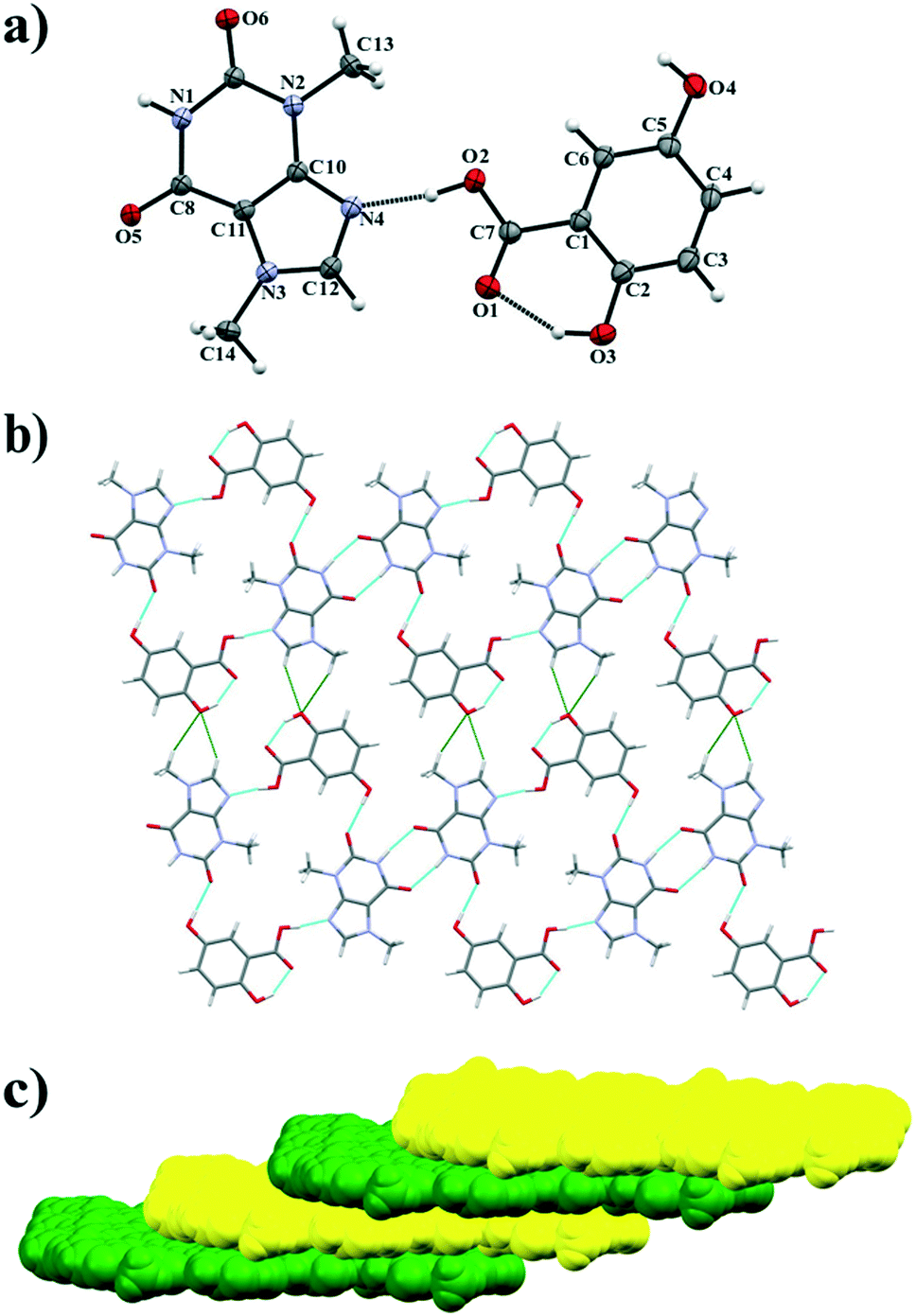
3.1.2. Theobromine-2,4-dihydroxybenzoic acid cocrystal.
TBR and 24DHBA form a cocrystal in a 1:1 stoichiometric ratio in the monoclinic P21/n space group. The asymmetric unit consists of one molecule of each component (Fig. 4a). TBR and 24DHBA form 1D linear polymer chains parallel to the (![[4 with combining macron]](https://www.rsc.org/images/entities/char_0034_0304.gif) 52) crystallographic plane (Fig. 4b). The hydrogen atom in the carboxyl group of 24DHBA is hydrogen bonded via O2–H2⋯N4 interaction with TBR (synthon B1 – Fig. 2, Table 2). The ortho-hydroxyl group in the acid participates in an intramolecular O3–H3⋯O1 hydrogen bond. The oxygen atom in the para-hydroxyl group of the acid accepts a proton from the N–H group in the pyrimidine ring of TBR (N1–H1⋯O4i interaction, synthon D1) and it is also a proton donor to the endo-carbonyl group of the TBR molecule (O4–H4⋯O6ii hydrogen bond, synthon C1) present in the neighboring chain parallel to the (
52) crystallographic plane (Fig. 4b). The hydrogen atom in the carboxyl group of 24DHBA is hydrogen bonded via O2–H2⋯N4 interaction with TBR (synthon B1 – Fig. 2, Table 2). The ortho-hydroxyl group in the acid participates in an intramolecular O3–H3⋯O1 hydrogen bond. The oxygen atom in the para-hydroxyl group of the acid accepts a proton from the N–H group in the pyrimidine ring of TBR (N1–H1⋯O4i interaction, synthon D1) and it is also a proton donor to the endo-carbonyl group of the TBR molecule (O4–H4⋯O6ii hydrogen bond, synthon C1) present in the neighboring chain parallel to the (![[6 with combining macron]](https://www.rsc.org/images/entities/char_0036_0304.gif)
![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) 8) crystallographic plane. Two sets of polymer chains are inclined by 62.294(3)° and they are stabilized through π(TBR)⋯π(24DHBA) forces (Fig. 4c, Table S1†). The TBR–24DHBA three-dimensional network is formed thanks to C–H⋯O interactions between adjacent stacks (Fig. 4d).
8) crystallographic plane. Two sets of polymer chains are inclined by 62.294(3)° and they are stabilized through π(TBR)⋯π(24DHBA) forces (Fig. 4c, Table S1†). The TBR–24DHBA three-dimensional network is formed thanks to C–H⋯O interactions between adjacent stacks (Fig. 4d).
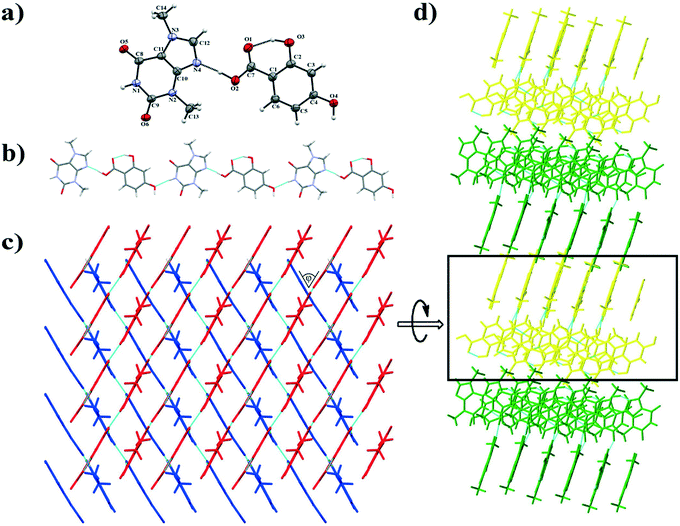 |
| | Fig. 4 a) ORTEP representation of the TBR·24DHBA asymmetric unit with numbering of atoms (thermal ellipsoids are plotted with the 50% probability level); b) polymer chain consisting of alternately hydrogen bonded TBR and 24DHBA molecules; c) 2D structure, viewed along the (101) crystallographic plane, composed of two sets of polymer chain stacks inclined by φ = 62.294(3)°; d) 3D structure of TBR·24DHBA held by C–H⋯O interactions. | |
3.1.3. Theobromine-2,5-dihydroxybenzoic acid cocrystal.
Theobromine-2,5-dihydroxybenzoic acid cocrystal crystallizes in the P space group in a 1:1 ratio. The asymmetric unit contains one acid and one alkaloid molecule (Fig. 5a). TBR and 25DHBA form molecular ribbons (Fig. 5b). Within the ribbons TBR molecules are held together by N1–H1⋯O5ii hydrogen bonds (synthon A2 – Fig. 2, Table 2) to form an R22(8) cyclic array. An imidazole nitrogen atom of TBR accepts the proton from the 25DHBA carboxyl group (O2–H2⋯N4 interaction, synthon B1). At the same time the meta-hydroxyl group in the acid is a proton donor to the oxygen atom in the endo-carbonyl group of TBR (O4–H4⋯O6i hydrogen bond, synthon C1). An intramolecular O3–H3⋯O1 hydrogen bond in the 25DHBA molecule forms an S11(6) motif. The TBR–25DHBA ribbons are connected by C–H⋯O interactions to form sheets parallel to the (212) crystallographic plane (Fig. 5b). Layers are stacked in an offset manner and are sustained by π(TBR)⋯π(25DHBA) forces (Fig. 5c, Table S1†).
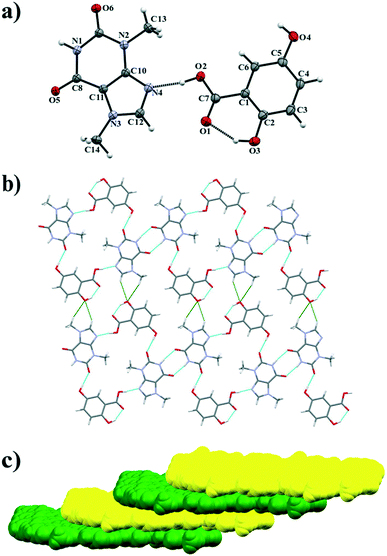 |
| | Fig. 5 a) ORTEP representation showing the asymmetric unit of the TBR·25DHBA cocrystal with numbering of atoms (thermal ellipsoids are drawn with the 50% probability level); b) molecular layer composed of 1D ribbons connected by C–H⋯O hydrogen bonds (marked in green colour); c) 3D network (“stair” motif) formed by TBR and 25DHBA molecules held by π-stacking interactions. | |
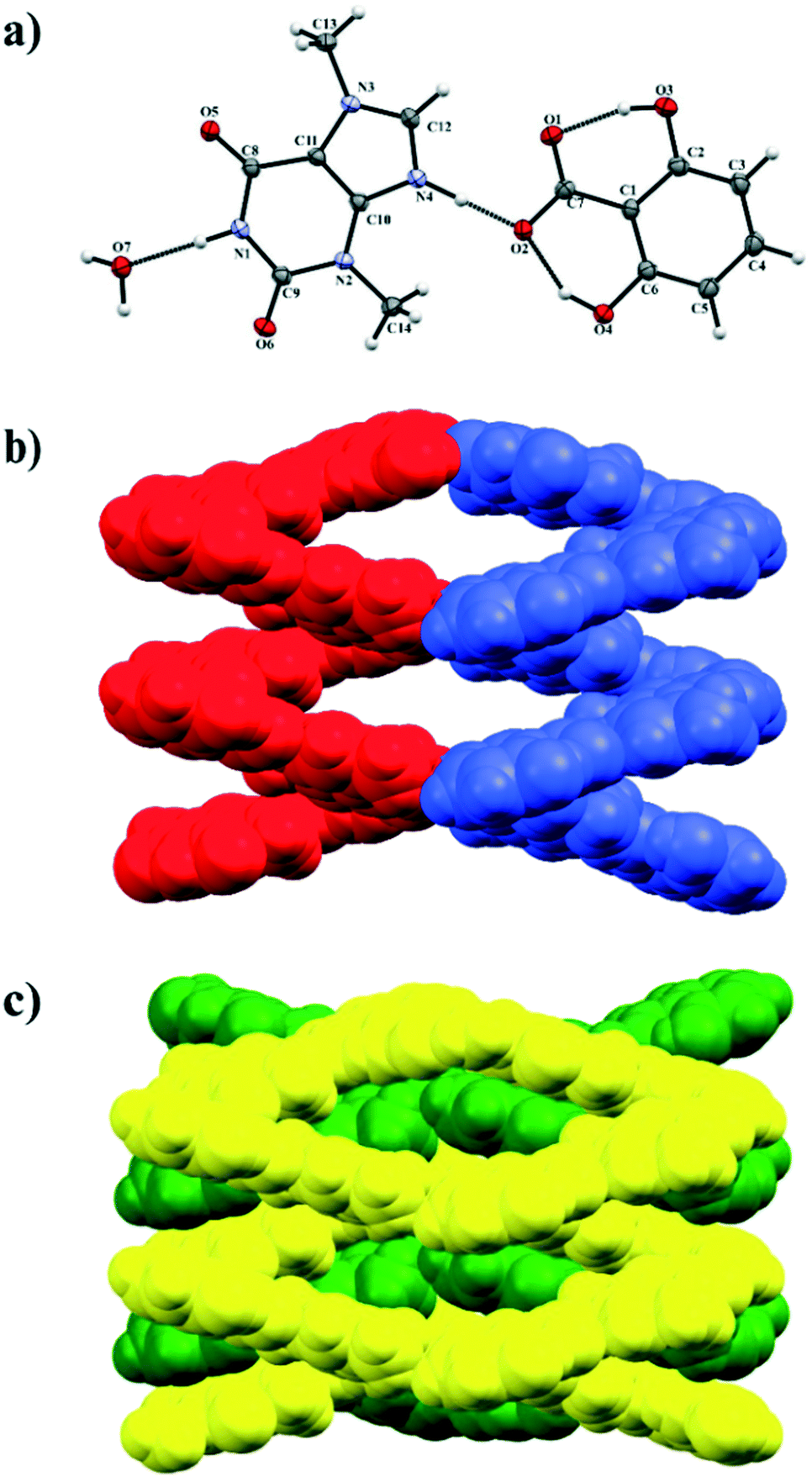
3.1.4. Theobrominium-2,6-dihydroxybenzoate monohydrate.
Theobromine and 2,6-dihydroxybenzoic acid cocrystallize as a monohydrate salt (TBR-H)+·(26DHBA)−·H2O. The crystal structure was solved in the monoclinic P21/n space group. The asymmetric unit consists of one (TBR-H)+ cation, one 2,6-dihydroxybenzoate anion and a water molecule (Fig. 6a). The structural analysis unambiguously indicated the proton position at the imidazole nitrogen atom of theobromine. In the 26DHBA molecule intramolecular charge assisted O–H⋯O− hydrogen bonds are observed (O3–H3⋯O1 and O4–H4A⋯O2) and they form a 2S11(6) motif. A (TBR-H)+ monocation is connected by an N4–H4⋯O2− hydrogen bond with an acid monoanion. The TBR molecule is hydrogen bonded with two solvent molecules via N1–H1⋯O7 (synthon D2, Fig. 2, Table 2) and O7–H7B⋯O5ii (synthon C3) interactions. At the same time one of the hydroxyl groups of the 26DHBA anion accepts a proton from a water molecule (O7–H7A⋯O4i hydrogen bond). In the crystal structure of (TBR-H)+·(26DHBA)−·H2O helices of opposite handedness are present and the solvent molecules are responsible for connecting adjacent helical systems (Fig. 6b). The left-handed helices extend along the 21 axis at the ¼, y, ¼ position and the right-handed helices extend along the 21 axis at the ¾, y, ¾ position. The pitch of these helices is equal to 6.845(1) Å, determined as a O7⋯O7x,y+1,z distance. Additionally, in the 3D network the opposite-handed helices are interdigitated and held together by C–H⋯O and π-stacking interactions (Fig. 6c, Table S1†).
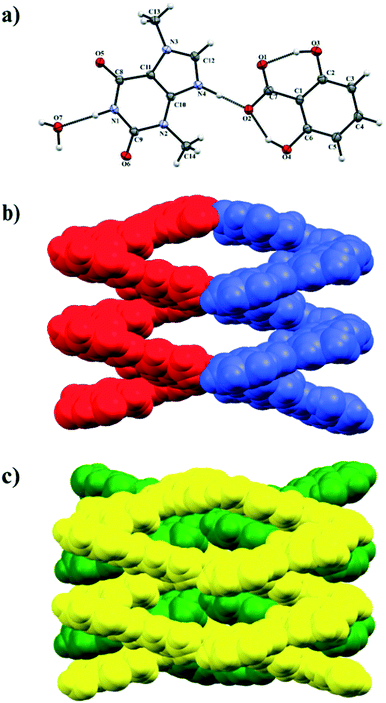 |
| | Fig. 6 a) ORTEP representation of the (TBR-H)+·(26DHBA)−·H2O asymmetric unit (thermal ellipsoids are drawn with the 50% probability level); b) opposite-handed helices connected by water molecules (red colour – left-handed helix along the 21 axis at the ¼, b, ¼ position, blue colour – right-handed helix along the 21 axis at the ¾, b, ¾ position; c) 3D structure formed by interdigitated helices held by stacking interactions. | |
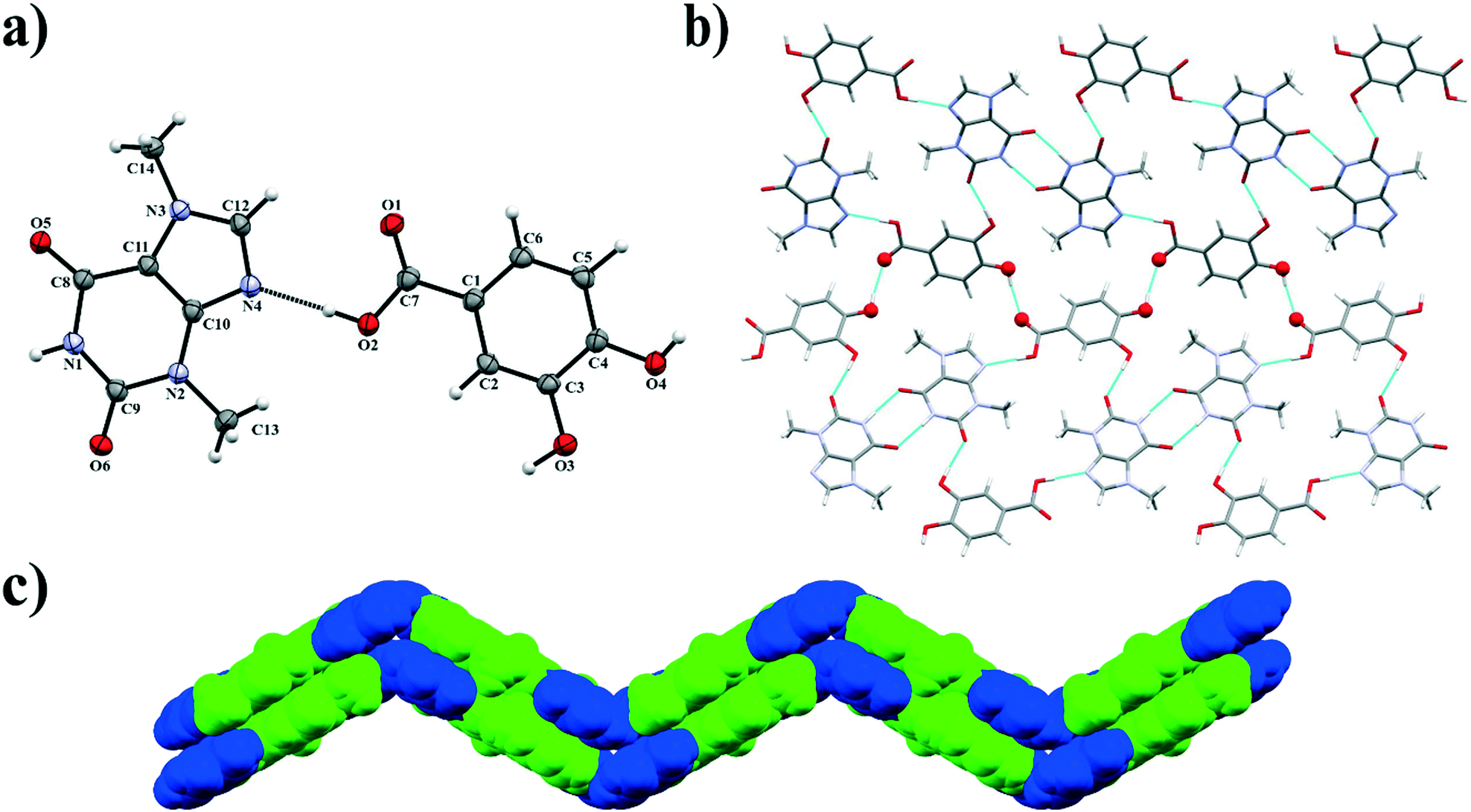
3.1.5. Theobromine-3,4-dihydroxybenzoic acid cocrystal.
Theobromine (TBR) and 3,4-dihydroxybenzoic acid (34DHBA) cocrystallize in the orthorhombic Pbcn space group in a stoichiometric ratio (1:1) with both one TBR and one 34DHBA molecule in the asymmetric unit (Fig. 7a). Components of this cocrystal form 1D ribbons parallel to the (201) and (20) crystallographic planes inclined by 120.6° (Fig. 6b). The equivalent 1D layers are at 13.376(1) Å from each other. In this system synthons R22(8) are formed between two TBR molecules through N1–H1⋯O5iii interactions (synthon A2 – Fig. 2, Table 2). Each alkaloid molecule is hydrogen bonded via Nimidazol⋯HOOC interaction with one 34DHBA (synthon B1) and with a second acid molecule via O–H(meta)⋯O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C(endo-carbonyl) interaction (synthon C1, Fig. 2). These ribbons are held together by O4–H4⋯O1ii hydrogen bonds between the carboxyl group in one acid and the para-hydroxyl group in the second acid molecule, resulting in “wavy” zigzag sheet formation (Fig. 7c). These 2D systems form stacks sustained by C–H⋯O forces and π(TBR)⋯π(TBR) and π(TBR)⋯π(34DHBA) interactions (Table S1†).
C(endo-carbonyl) interaction (synthon C1, Fig. 2). These ribbons are held together by O4–H4⋯O1ii hydrogen bonds between the carboxyl group in one acid and the para-hydroxyl group in the second acid molecule, resulting in “wavy” zigzag sheet formation (Fig. 7c). These 2D systems form stacks sustained by C–H⋯O forces and π(TBR)⋯π(TBR) and π(TBR)⋯π(34DHBA) interactions (Table S1†).
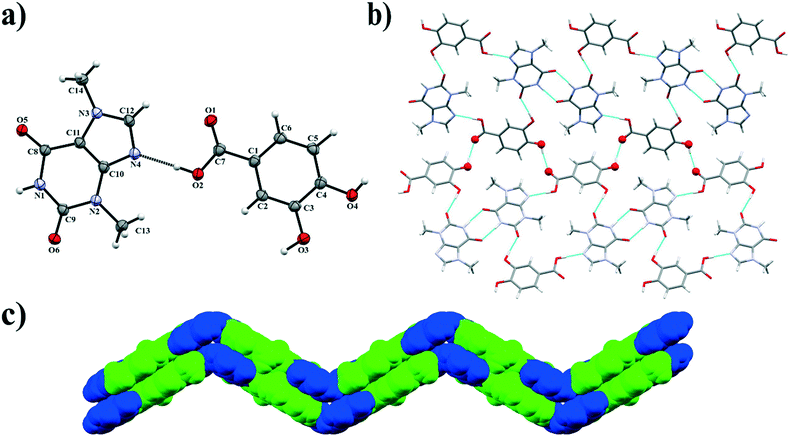 |
| | Fig. 7 a) ORTEP representation of the TBR·34DHBA cocrystal with an atomic numbering scheme (thermal ellipsoids are drawn with the 50% probability level); b) 2D structure composed of molecular ribbons connected by O–H⋯O hydrogen bonds present on the sheet bend; c) the “zigzag” sheet formed by π-stacking interactions between 1D ribbons (blue and green colours represent 34DHBA and TBR molecules, respectively). | |
3.1.6. Theobromine-3,5-dihydroxybenzoic acid cocrystal.
The crystal structure of theobromine and 3,5-dihydroxybenzoic acid in 1:1 stoichiometry was solved in the monoclinic P21/c space group. There are two TBR and two 35DHBA molecules in the asymmetric unit. These components form two-dimensional layers parallel to the (101) crystallographic plane. Two alkaloid molecules form a dimer through N–H⋯O hydrogen bonds (synthon A1) and the result is the cyclic array R22(8) formation. Each TBR molecule is connected with two 35DHBA molecules by O–H⋯N (synthon A2) and O–H⋯O (synthon B1) hydrogen bonds, respectively. The second hydroxyl group from the acid is involved in dimer acid–acid R22(14) formation by CO(carboxyl)⋯H–O(hydroxyl) interactions. In this cocrystal the two hydroxyl groups of the acid molecules adopt the most favorable anti–anti conformation.76 The TBR–35DHBA sheets form stacks along [010] and they are held together by π(TBR)⋯π(35DHBA) interactions (Fig. 8c, Table S1†).
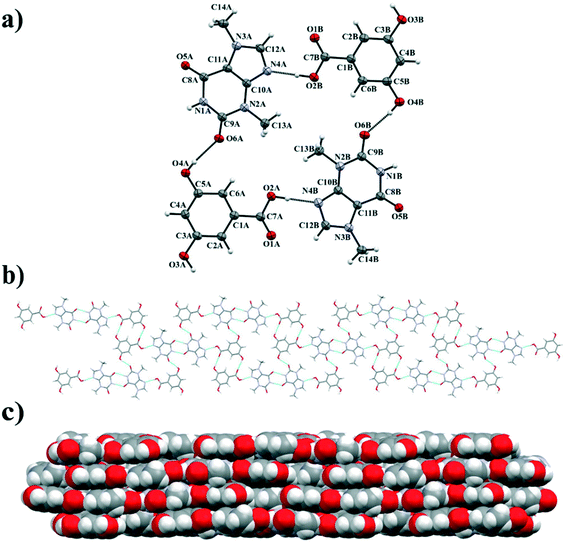 |
| | Fig. 8 a) ORTEP representation of the asymmetric unit in the TBR·35DHBA cocrystal (thermal ellipsoids are plotted with the 50% probability level); b) the 2D molecular layer consisting of TBR and 35DHBA molecules; c) the 3D structure composed of layers sustained by π-stacking forces. | |
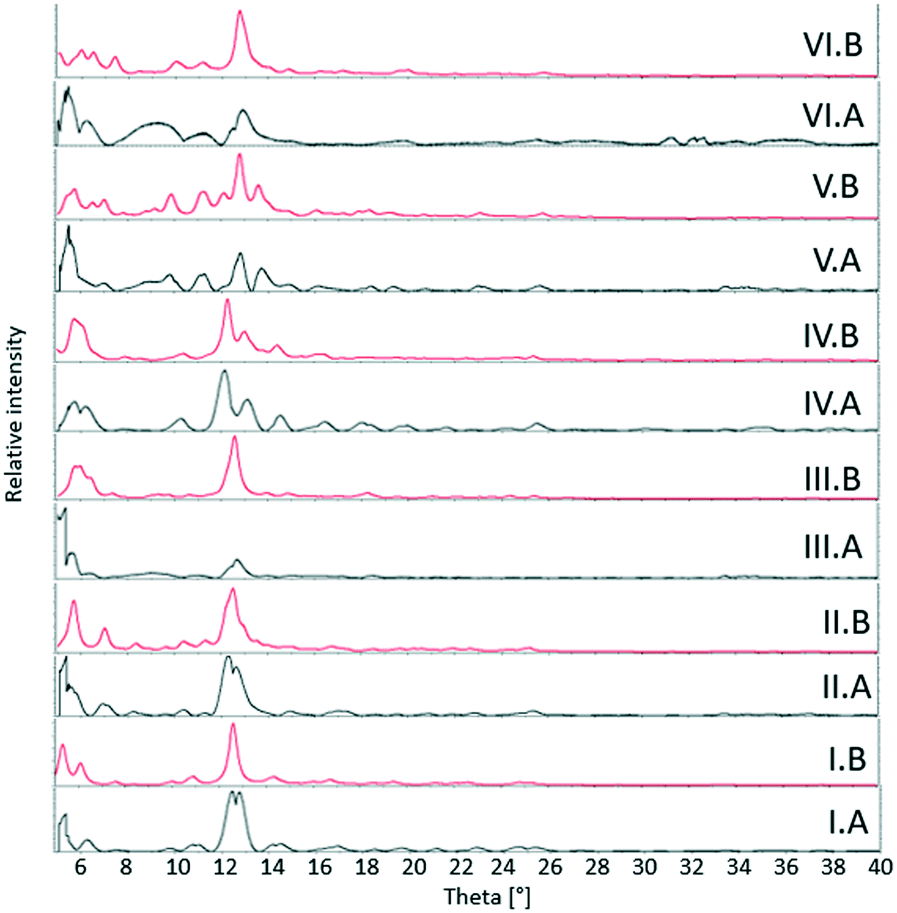
3.2. Powder X-ray diffraction
The theoretical powder XRD patterns for theobromine–dihydroxybenzoic acid systems were generated using the Mercury program.71 They were compared with the powder XRD patterns for the samples from neat grinding (II – TBR·24DHBA, III – TBR·25DHBA, V – TBR·34DHBA, VI – TBR·35DHBA) and liquid-assisted grinding (I – TBR·23DHBA·H2O and IV – (TBR-H)+·(26DHBA)−·H2O). The experimental and theoretical diffractograms denoted by A and B, respectively, for I, II, III, IV and V samples are similar. We can see differences in the peak intensity for the two powder XRD patterns of III and V substances. For cocrystal VI, differences in both powder diffractograms are visible, particularly in the peak positions and intensities, which can be explained by the difference in the degree of sample crystallinity (Fig. 9).
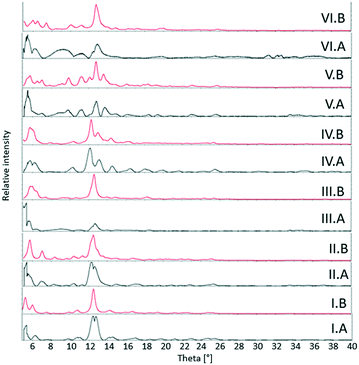 |
| | Fig. 9 Powder XRD patterns for described theobromine systems. I – TBR·23DHBA·H2O, II – TBR·24DHBA, III – TBR·25DHBA, IV – (TBR-H)+·(26DHBA)−·H2O, V – TBR·34DHBA, and VI – TBR·35DHBA. A – powder XRD patterns for samples from grinding, B – theoretical powder XRD patterns generated from the single crystal structure. | |
3.3. Solubility measurements
The current great attention towards development of cocrystals is due to the ability of cocrystals to fine tune the solubility properties of APIs. Theobromine is very slightly soluble in water (0.330 g L−1).29 Therefore, to improve its aqueous solubility cocrystals with dihydroxybenzoic acids were prepared. Solubility values were determined using steady-state absorption spectroscopy.
The results summarized in Table 5 show an improved solubility in all six solids compared to the pure TBR. The solubility of cocrystals with dihydroxybenzoic acid containing ortho-hydroxyl groups increases with increasing coformer solubility. The exception is (TBR-H)+·(26DHBA)−·H2O, which is the most highly soluble complex among all the cocrystals described in this paper and shows 100 times improvement in the solubility of TBR. TBR·25DHBA is the least soluble cocrystal, whose solubility is 4.4 times higher than the solubility of TBR. The solubilities of 34DHBA and 35DHBA are comparable. Surprisingly, TBR·35DHBA shows an about 26 fold increase in the solubility of TBR whereas for TBR·34DHBA only 7 times.
Table 5 Solubility of theobromine cocrystals in water. The increase relative to TBR is shown in parentheses
|
|
Absorption solubilitya (g L−1) |
Aqueous solubility of coformers (g L−1) |
|
The values in parentheses indicate the extent of increase (×) relative to the solubility of TBR.
|
| TBR·23DHBA·H2O |
6.25 (×18.9) |
29.1 (ref. 77) |
| TBR·24DHBA |
1.60 (×12.2) |
8.0 (ref. 78) |
| TBR·25DHBA |
1.45 (×4.4) |
2.2 (ref. 79) |
| (TBR-H)+·(26DHBA)−·H2O |
33.3 (×100) |
9.56 (ref. 80) |
| TBR·34DHBA |
2.20 (×6.7) |
12.4 (ref. 81) |
| TBR·35DHBA |
8.76 (×26.5) |
12.0 (ref. 82) |
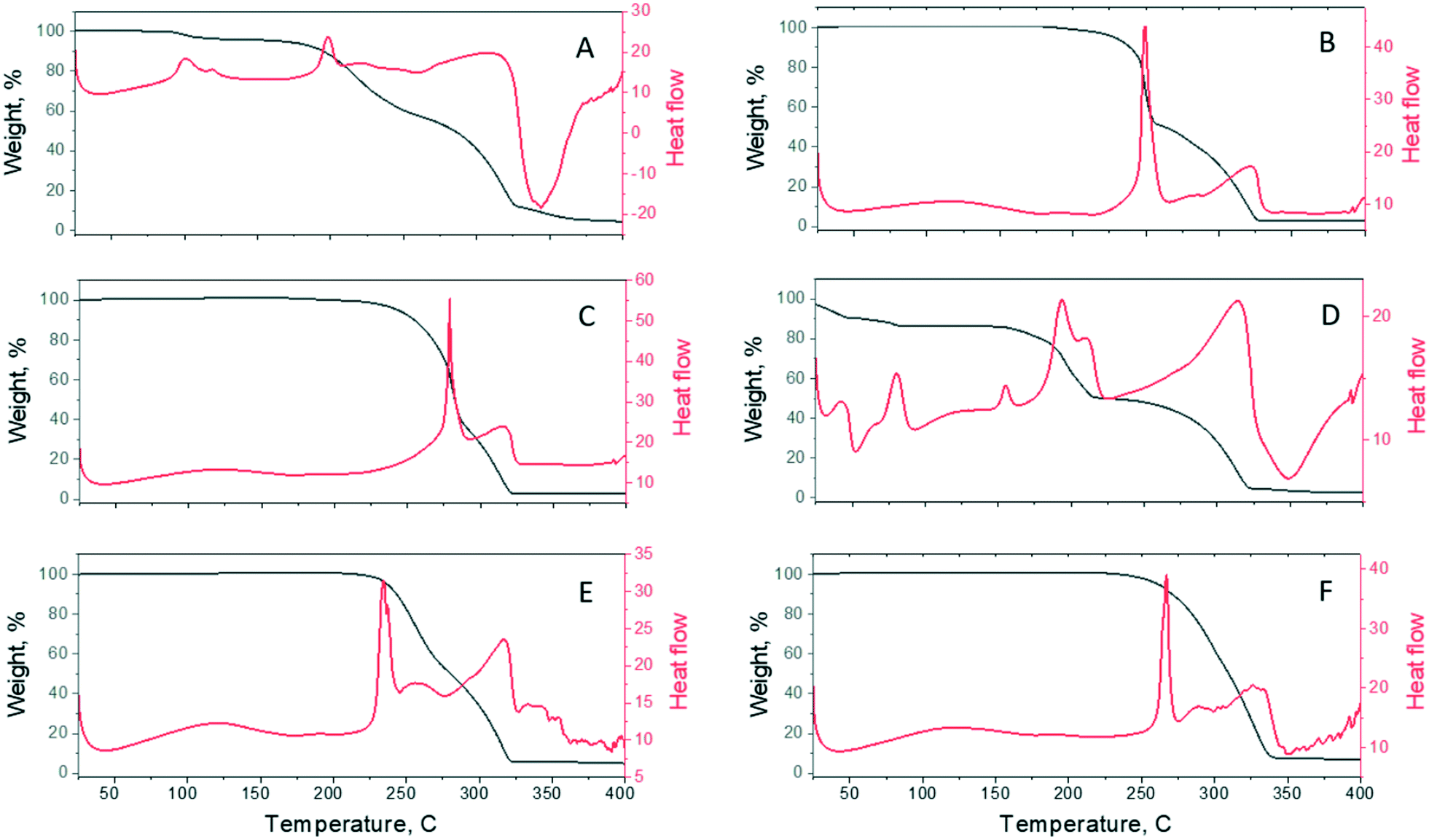
3.4. Simultaneous thermal analysis (STA)
The thermal behaviour of the resulting cocrystals was characterized by DSC, as shown in Fig. 10. Four out of the six (TBR·24DHBA, TBR·25DHBA, TBR·35DHBA, and TBR·34DHBA) samples display a sharp melting endotherm, indicating highly crystalline materials. In their DSC curves, two signals are observed. The first signal refers to the complete decomposition of appropriate dihydroxybenzoic acid. The second signal, around 310–320 °C, is attributed to the decomposition of the TBR molecules. The melting points of the cocrystals and starting materials are tabulated in Table 6. The melting points of the cocrystals range from 198 to 279 °C although all the cocrystals are position isomers. There is no correlation between the melting point of the cocrystals and the melting point of the coformers but cocrystal melting points are between those of the coformer and theobromine. Since the decomposition temperature of dihydroxybenzoic acids is higher than that of the pure coformers, it can be concluded that the 24DHBA, 25DHBA, 34DHBA and 35DHBA molecules are stabilized in the cocrystals.
Table 6 Melting points of cocrystals and coformers
|
|
Coformer melting point (°C) (ref. 83) |
Cocrystal melting point (°C) |
| 23DHBA |
208 |
198 |
| 24DHBA |
225 |
250 |
| 25DHBA |
201 |
279 |
| 34DHBA |
201 |
235 |
| 35DHBA |
238 |
268 |
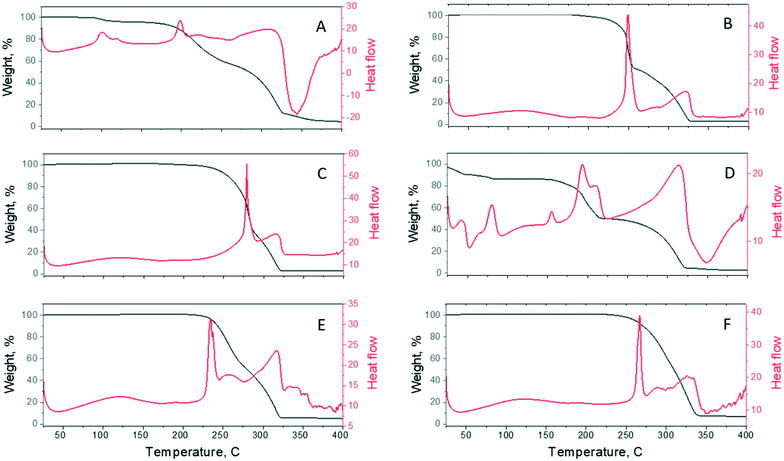 |
| | Fig. 10 The simultaneous thermal analysis (STA) curves of A) TBR·23DHBA·H2O, B) TBR·24DHBA, C) TBR·25DHBA, D) (TBR-H)+·(26DHBA)−·H2O, E) TBR·34DHBA, and F) TBR·35DHBA. TGA and DSC curves are presented by black and red colours, respectively. | |
The presence of water in the TBR·23DHBA·H2O crystal structure is evident from the DSC measurement. The signals around 100 °C are connected with the release of the water molecules from the crystal structure. Based on the weight loss in the 90–130 °C temperature range, the percentage of water content is equal to about 5%. This value is in agreement with single crystal X-ray analysis. Below 90 °C no weight loss is observed, which leads to the conclusion that there is no unbound water in the sample. The next two signals at 198 °C and 310 °C refer to complete decomposition of 23DHBA and TBR molecules, respectively.
The (TBR-H)+·(26DHBA)−·H2O salt exhibits a melting point at 193 °C, between those of TBR (351 °C)72 and 26DHBA (173 °C).83 For this salt, the DSC endotherm shows first signals below 100 °C. They can be connected with the release of water molecules. In the 100–250 °C temperature range, three endothermic signals are observed. In this range, the material loss reaches 32%. It can be concluded that the signals refer to complete decomposition of 26DHBA. There is no sharp melting endotherm, which excludes the presence of crystalline parts. The signal at 313 °C is attributed to the decomposition of the TBR molecules.
4. Discussion
Eight of 32 theobromine structures deposited in the CSD83 (without repeats and RUTHEV49) contain theobromine–carboxylic acid systems: CSATBR,48 GORGUR,50 HIJYAB,51 HIJYEF,51 MUPPET,55 NURYUV,52 ZIZRUX49 (the same as ZIZRUX01 (ref. 53)) and ZOYBOG.57 Additionally, TBR·2HBA, TBR·3HBA and TBR·4HBA·H2O complexes were taken into consideration for supramolecular synthon analysis of theobromine.42
4.1. Synthon hierarchy – synthon A1 vs. synthon A2 (homosynthon amide–amide)
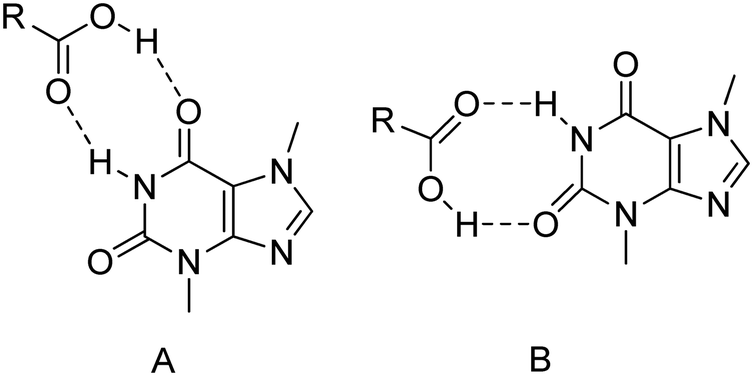
The theobromine molecule, in contrast to theophylline and caffeine, can form two types of amide–amide homosynthons depending on the endo- or exo-carbonyl oxygen atom, which is involved in it (synthons A1 and A2, Fig. 2). In TBR·2HBA, TBR·3HBA, TBR·25DHBA, TBR·34DHBA and TBR·35DHBA cocrystals, TBR–TBR homosynthons are formed through the exo-oxygen atom (synthon A2). A homodimer with endo-oxygen atom participation (synthon A1) is only observed in the TBR·4HBA·H2O complex. In TBR·23DHBA·H2O, TBR·24DHBA and (TBR-H)+·(26DHBA)−·H2O, an amide–amide motif is not present. Synthon A2 TBR–TBR is more favourable than synthon A1 in this alkaloid cocrystal within carboxylic acids. For comparison, in the crystal lattice of pure theobromine, synthon II is present (SEDNAQ).84 Our results are in line with deposited structures in the CSD.84 In four structures, this kind of homosynthon is formed by the exo-carbonyl oxygen atom (GORGUR, HIJYAB, NURYUV, and ZIZRUX). In the remaining four entries (CSATBR, HIJYEF, MUPPET, and ZOYBOG), homosynthons TBR–TBR are not observed, but two types of heterosynthons amide–acid are present (Fig. 11). In CSATBR, the exo-oxygen atom and in HIJYEF, MUPPET and ZIZRUX, the endo-oxygen atom take part in this heterodimer formation.
 |
| | Fig. 11 Two possible types of amide–carboxylic acid heterosynthons in theobromine–carboxylic acid systems. | |
4.2. Synthon hierarchy – synthon B1 vs. synthon B2 (heterosynthon with imidazole nitrogen atom participation)
In 6 out of the 8 entries in the CSD, the imidazole nitrogen atom of theobromine accepts a proton from the carboxyl group (COOH⋯Nimidazole hydrogen bond, Fig. 2). In all of the theobromine–dihydroxybenzoic acid systems, the same interaction (synthon B1) is present. When the theobromine forms cocrystals with 2HBA and 3HBA, the same motif is formed. Only in the TBR·4HBA·H2O cocrystal hydrate is that the hydroxyl group is a proton donor to the Nimidazole atom (synthon B2, Fig. 2, Table 7), because pairs of carboxylic acid molecules form homosynthons. The carboxylic acid–carboxylic acid homosynthon formation is rare in the closed neighbourhood of the alkaline N(aromatic) atom, and thus is interesting.43 In caffeine cocrystals with hydroxybenzoic acids as coformers, only an Nimidazole⋯HOOC synthon is formed (Table 9) and acid–acid homodimers are not present.44,87 However, in theophylline cocrystals with 2HBA, 3HBA, 25DHBA and 35DHBA, synthon B1 is present. In theophylline cocrystals with 4HBA, 23DHBA, 24DHBA and 34DHBA (polymorph II), theophylline–acid heterosynthons are formed through N–H⋯OCcarboxyl and COexo⋯H–Ocarboxyl hydrogen bonds (Table 8) and the hydroxyl group is a donor proton for the imidazole nitrogen atom (synthon B2). In polymorph I of TPH·34DHBA, two carboxyl groups are engaged in acid–acid homodimer formation, thus the remaining two hydroxyl groups compete in proton donation to the Nimidazole atom (the meta-hydroxyl group is a donor proton for the endo-oxygen atom in TPH, so only the para-hydroxyl group can donate a proton to the imidazole nitrogen atom).43,85,86
Table 7 Summary of the contribution of particular theobromine groups to supramolecular synthon formation in theobromine cocrystals with mono- and dihydroxybenzoic acids
| TBR complex |
Nimidazole |
COexo |
N–Hpyrimidine |
COendo |
| TBR·2HBA |
B1 |
A2 |
A2 |
× |
| TBR·3HBA |
B1 |
A2 |
A2 |
C1 |
| TBR·2(4HBA)·H2O |
B2 |
C3 |
A1 |
A1, C2 |
| TBR·23DHBA·H2O |
B1 |
× |
D1 |
C2 |
| TBR·24DHBA |
B1 |
× |
D1 |
C1 |
| TBR·25DHBA |
B1 |
A2 |
A2 |
C1 |
| (TBR-H)+·(26DHBA)−·H2O |
Proton transfer |
C3 |
D2 |
× |
| TBR·34DHBA |
B1 |
A2 |
A2 |
C1 |
| TBR·35DHBA |
B1 |
A2 |
A2 |
C1 |
Table 8 Supramolecular synthons in theophylline cocrystals with mono-42 and dihydroxybenzoic acids43,85,86
| TPH complex |
REFCOD |
Nimidazole |
N–Himidazole |
COexo |
COendo |
|
TPH–TPH homosynthons formed through N–H⋯OCexo hydrogen bonds.
TPH–acid heterosynthons formed through N–H⋯OCcarboxyl and COexo⋯H–Ocarboxyl hydrogen bonds.
|
| TPH·2HBAa |
KIGLES |
COOH⋯Nimidazole |
N–H⋯OCexo |
COexo⋯H–Nimidazole |
× |
| TPH·3HBAa |
DOPMUS |
COOH⋯Nimidazole |
N–H⋯OCexo |
COexo⋯H–Nimidazole |
COendo⋯H–Ohydroxyl |
| TPH·4HBAb |
KIGLOC |
OH⋯Nimidazole |
N–H⋯OCcarboxyl |
COexo⋯H–Ocarboxyl |
× |
| TPH·23DHBAb |
DOPNAZ |
OH⋯Nimidazole |
N–H⋯OCcarboxyl |
COexo⋯H–Ocarboxyl |
× |
| TPH·24DHBAb |
DOPNED |
OH⋯Nimidazole |
N–H⋯OCcarboxyl |
COexo⋯H–Ocarboxyl |
COendo⋯H–Ohydroxyl |
| TPH·24DHBA·H2Ob |
DEYREF |
HO–H⋯Nimidazole |
N–H⋯OCcarboxyl |
COexo⋯H–Ocarboxyl |
COendo⋯H–OH |
| TPH·25DHBAa |
DUCROJ |
COOH⋯Nimidazole |
N–H⋯OCexo |
COexo⋯H–Nimidazole |
COendo⋯H–Ohydroxyl |
| (TPH-H)+·(26DHBA)−·H2O |
WOCHED01 |
COO−⋯H–Nimidazole |
N–H⋯OH2 |
COexo⋯H–OH |
× |
| TPH·34DHBA (polymorph I)a |
WOCHON |
OH⋯Nimidazole |
N–H⋯OCexo |
COexo⋯H–Nimidazole |
COendo⋯H–Ohydroxyl |
| TPH·34DHBA (polymorph II)b |
WOCHON02 |
OH⋯Nimidazole |
N–H⋯OCcarboxyl |
COexo⋯H–Ocarboxyl |
COendo⋯H–Ohydroxyl |
| TPH·35DHBAa |
WOCHIH01 |
COOH⋯Nimidazole |
N–H⋯OCexo |
COexo⋯H–Nimidazole |
COendo⋯H–Ohydroxyl |
Additionally, in two theobromine–acid structures found in the CSD (MUPPET and ZOYBOG), the imidazole nitrogen atom of theobromine accepts a proton from a water molecule (Nimidazole⋯H–OH synthon, Fig. 12).83 This synthon, which is interesting, was also found only in two alkaloid–acid systems, i.e. in theophylline–2,4-dihydroxybenzoic acid monohydrate (DEYREF) and caffeine–3,4,5-tri hydroxybenzoic acid hexahydrate (ZICGIE).84
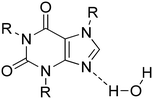 |
| | Fig. 12 The heterosynthon formed between a water molecule and the imidazole nitrogen atom of the alkaloid molecule. | |
4.3. Synthon hierarchy – synthons C1, C2 and C3 (heterosynthons with exo- and endo-carbonyl group participation)
The exo- and endo-oxygen atoms in the theobromine pyrimidine ring can take part in amide–amide homosynthon (observed in the structure in our work) or in amide–acid heterosynthon formation. Moreover, near these synthons there is another oxygen atom, which can also be a potential acceptor of protons. In theobromine cocrystals with 3HBA, 25DHBA, 34DHBA and 35DHBA, where homosynthon TBR–TBR with an exo-oxygen atom is present, endo-oxygen atoms are proton acceptors from hydroxyl groups (synthon C1, Fig. 2). A similar situation is found in ZIZRUX, where the endo-carbonyl group is a proton acceptor from the amine group. In the TBR·2HBA cocrystal (and in GORGUR, HIJYAB, and NURYUV) with the same TBR–TBR homodimer, the endo-oxygen atom does not take part in strong hydrogen bond formation. In TBR·4HBA·H2O with TBR–TBR homosynthons formed through endo-oxygen atoms, water molecules are proton donors to both exo- and endo-oxygen atoms (synthons C2 and C3). In TBR·23DHBA·H2O, TBR·24DHBA and (TBR-H)+·(26DHBA)−·H2O, TBR–TBR homosynthons are not formed. In TBR·23DHBA·H2O, the endo-oxygen atom is a double proton acceptor from two water molecules (synthon C2), in salt monohydrate with 26DHBA, the exo-oxygen atom is a single proton acceptor from a water molecule (synthon C3), and in TBR·24DHBA, the endo-oxygen atom accepts a proton from the hydroxyl group of the acid (synthon C1).
In CSATBR, where acid–amide heterosynthons are formed through the exo-oxygen atom, the endo-oxygen atom does not take part in strong hydrogen bond formation. There are also three structures with amide–acid heterosynthons with exo-carbonyl group participation. In HIJYEF, the exo-oxygen atom is not hydrogen bonded, in MUPPET, this oxygen atom is a double proton acceptor from two water molecules, and in ZOYBOG, the endo-oxygen atom accepts a proton from the hydroxyl group, and, additionally, water molecules are proton donors to the exo-oxygen atom.
4.4. Synthons D1 and D2 (with an amine group in pyrimidine ring participation)
In theobromine–acid systems deposited in the CSD, the N–H group of the theobromine pyrimidine ring takes part in the formation of either amide–amide homosynthons or amide–acid heterosynthons.84 From the nine theobromine complexes with mono- and dihydroxybenzoic acids in TBR·23DHBA·H2O, TBR·24DHBA and (TBR-H)+·(26DHBA)−·H2O, homosynthon amide–amide is not formed. In the first two systems, N–H groups are proton donors for oxygen atoms in meta- or para-hydroxyl groups, respectively (synthon D1, Fig. 2, Table 7). In the crystal structure of the salt containing a 2,6-dihydroxybenzoate anion, the N–H group is a proton donor to the oxygen atom of water molecules (synthon D2).
4.5. Hydrate formation
Theobromine cocrystallizes as a monohydrate with 4HBA, 23DHBA and 26DHBA as coformers. In theophylline cocrystal analogues, hydrates are formed with 24DHBA85 and 26DHBA.43 Four monohydrates of caffeine with 4HBA,87 23DHBA,44 24DHBA44 and 35DHBA44 as coformers have been published. The role of water molecules in the crystal lattice may be the balanced ratio of hydrogen-donors to the number of hydrogen-acceptors.88 However, in the case of alkaloid cocrystals with mono- and dihydroxybenzoic acids, the influence of the steric effects is more significant in hydrate formation. In the TBR·4HBA·H2O hydrate, while two carboxyl groups form a homodimer and one hydroxyl group of one acid is a proton donor to the imidazole nitrogen atom, a second hydroxyl group is connected with the theobromine molecule by a water molecule to minimize contacts between benzene (4HBA) and the methyl group near the pyrimidine ring. In TBR·23DHBA·H2O, the meta-hydroxyl group of one acid molecule accepts a proton from the N–H group in the pyrimidine ring of TBR, so direct connection of the meta-hydroxyl group in the second acid molecule and the endo-oxygen atom of the same theobromine molecule could cause repulsive interactions between two benzene rings of 23DHBA. In (TBR-H)+·(26DHBA)−·H2O, water molecules localized between two theobromine molecules decrease repulsive interactions between methyl groups near pyrimidine and benzene (26DHBA) rings. This is the reason why TBR–TBR homosynthons in the crystal lattice of this complex are not formed. This example is similar to the (TPH-H)+·(26DHBA)−·H2O hydrate, where water is present between two theophylline molecules and homosynthons TPH–TPH are not present. In caffeine molecules, the methyl groups in the vicinity of endo- and exo-oxygen atoms provide steric hindrance, which destabilizes the crystal structure. Water molecules present in caffeine cocrystal hydrates play a crucial role in elimination of repulsive interactions between the aromatic ring in the acid and methyl groups by forming O–H⋯O hydrogen bonds. In CAF·4HBA·H2O, CAF·23HBA·H2O, CAF·24HBA·H2O and CAF·35HBA·H2O, water molecules are proton donors only to the exo-oxygen atom, while in CAF·4HBA·H2O and CAF·24HBA·H2O, each of them (endo- and exo-oxygen atoms) is a proton acceptor from different water molecules44,87 (Table 9). Additionally, Table 10 shows in which way water molecules affect the dimensionality of alkaloid–hydroxybenzoic acid hydrates.
Table 9 Supramolecular synthons in caffeine derivatives with mono- and dihydroxybenzoic acids44,87
| CAF complex |
REFCOD |
Nimidazole |
COexo |
COendo |
|
Hydrogen atoms in water molecules were not locatable on the difference Fourier map.
|
| CAF·2HBA |
XOBCAT |
COOH⋯Nimidazole |
× |
× |
| CAF·3HBA |
MOZCOU |
COOH⋯Nimidazole |
× |
COendo⋯H–Ohydroxyl |
| CAF·2(4HBA) |
MOZDAH |
COOH⋯Nimidazole |
COexo⋯H–Ohydroxyl |
COendo⋯H–Ohydroxyl |
| 2CAF·4HBA |
MOZCUA01 |
COOH⋯Nimidazole |
× |
COendo⋯H–Ohydroxyl |
| CAF·4HBA·H2O |
LATBIT |
COOH⋯Nimidazole |
COexo⋯H–OH |
COendo⋯H–OH |
| CAF·23DHBA·H2O |
MOZDEL |
COOH⋯Nimidazole |
COexo⋯H–OHa |
× |
| CAF·24DHBA·H2O |
MOZCIO |
COOH⋯Nimidazole |
COexo⋯H–OH |
COendo⋯H–OH |
| CAF·25DHBA |
MOZDIP |
COOH⋯Nimidazole |
COexo⋯H–Ohydroxyl |
× |
| CAF·35DHBA·H2O |
MOZCEK |
COOH⋯Nimidazole |
COexo⋯H–OH |
COendo⋯H–Ohydroxyl |
Table 10 The comparison of systems composed of strong hydrogen bonds in theobromine, theophylline and caffeine complexes with mono- and dihydroxybenzoic acids42–44,85–87
|
|
TBR |
TPH |
CAF |
|
Infinite system with water molecule participation.
CAF·26DHBA complex not found in the literature and in the CSD.84
Cocrystal was obtained, which was confirmed by powder XRD patterns, but cocrystallization of these substances did not give good quality monocrystals.44
|
| 2HBA |
Discrete |
1D infinite |
Discrete |
| 3HBA |
1D infinite |
1D infinite |
Discrete |
| 4HBA |
1D infinitea |
1D infinite |
MOZDAH – 2D infinite |
| MOZCUA01 – discrete |
| LATBIT – 2D infinitea |
| 23DHBA |
2D infinitea |
1D infinite |
1D infinitea |
| 24DHBA |
2D infinite |
DOPNED – 2D infinite |
2D infinitea |
| DEYREF – 2D infinitea |
| 25DHBA |
1D infinite |
2D infinite |
1D infinite |
| 26DHBA |
2D infinitea |
1D infinitea |
×b |
| 34DHBA |
2D infinite |
WOCHON – 2D infinite |
×c |
| WOCHON02 – 2D infinite |
| 35DHBA |
2D infinite |
2D infinite |
3D infinitea |
4.6. The ortho-hydroxyl groups form intra- and intermolecular hydrogen bonds
The ortho-hydroxyl groups in 2HBA, 23DHBA, 24DHBA, 25DHBA and 26DHBA in theobromine,42 theophylline43,85,86 and caffeine44,87 cocrystals form intramolecular hydrogen bonds O–Hhydroxyl⋯OCcarboxyl. This observation is compatible with one of the principles given by Etter:75
“6-membered-ring intramolecular hydrogen bonds form in preference to intermolecular hydrogen bonds.”
Therefore, the participation of this group in the intermolecular hydrogen bond formation was doubtful.89 An exception is the structure of (TBR-H)+·(26DHBA)−·H2O where one hydroxyl group is also a proton acceptor from a water molecule. The same situation is found in the CAF·23HBA·H2O cocrystal hydrate.44 The ortho-hydroxyl groups in TPH·2HBA and TPH·25HBA cocrystals form intermolecular O–Hhydroxyl⋯OCcarboxyl hydrogen bonds together with the carboxyl group in the neighboring acid molecule. The imidazole nitrogen atom in TPH·24HBA is a proton acceptor from the o-hydroxyl group.43 The fact of intermolecular hydrogen bond formation by ortho-hydroxyl groups in TBR, TPH and CAF cocrystals is difficult to explain so far and research in this area is ongoing.43
5. Conclusions
We prepared six new theobromine derivatives. Four theobromine cocrystals with 2,4-dihydroxy-, 2,5-dihydroxy, 3,4-dihydroxy and 3,5-dihydroxybenzoic acids, one TBR·23DHBA·H2O cocrystal hydrate and one salt hydrate containing a theobrominium cation and 2,6-dihydroxybenzoate anion were obtained by slow evaporation from solution and they were characterized by a single X-ray diffraction method. The powder XRD patterns confirmed the possibility of synthesis of these complexes by neat or liquid-assisted grinding. The UV-vis spectral measurements showed the improvement of theobromine solubility in water after cocrystallization. For neutral complexes, with no proton transfer, a 4.4- to 26.5-fold improvement in solubility compared to pure theobromine was demonstrated. For the salt monohydrate (TBR-H)+·(26DHBA)−·H2O, there is a 100-fold improvement in the theobromine solubility in water in relation to the pure alkaloid. For five theobromine cocrystals, the simultaneous thermal analysis showed an improvement in thermal stability after cocrystallization. Four samples (TBR·24DHBA, TBR·25DHBA, TBR·34DHBA and TBR·35DHBA) display a sharp melting endotherm, indicating highly crystalline materials. A lower melting endotherm is uncommon in cocrystals but reports for low melting-point cocrystals are available in the literature.90–92
In this paper, supramolecular analysis with homo- and heterosynthons responsible for self-organizing molecules in theobromine solids with mono- and dihydroxybenzoic acids was presented. Homosynthon amide–amide between two theobromine molecules with an exo-oxygen atom is more favorable than that with endo-oxygen atom participation. In most cases, the oxygen atoms not involved in TBR–TBR homodimer formation are proton acceptors from the hydroxyl group or water molecule(s). The acid–acid homosynthon occurs only in the TBR·4HBA·H2O cocrystal hydrate. In the intermolecular O–H⋯Nimidazole hydrogen bond, the carboxyl group is more often a proton donor than the hydroxyl group. In each solid, where the coformer has an ortho-hydroxyl group, an intramolecular O–H⋯O hydrogen bond is formed. Additionally, in (TPH-H)+·(26DHBA)−·H2O, one ortho-hydroxyl group accepts a proton from a water molecule. The pKa values of coformers do not affect the formation of particular supramolecular synthons by theobromine. These conclusions show how difficult it is to design a cocrystal structure from molecules containing many hydrogen-bonding groups. Our studies are in line with the trend of structural research and supramolecular synthon hierarchy in organic cocrystals. The similarities and differences in the formation of specific synthons presented in this paper are certainly important information in the topic of preferred synthons, not only in purine alkaloid cocrystals, indicating the need for further research in the field of organic cocrystal design.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by grant no. POWR.03.02.00-00-I026/16 co-financed by the European Union through the European Social Fund under the Operational Program Knowledge Education Development.
References
- K. Yuvaraja and J. Khanam, J. Pharm. Biomed. Anal., 2014, 96, 10–20 CrossRef CAS PubMed.
- V. P. Shah and G. L. Amidon, AAPS J., 2014, 16, 894–898 CrossRef CAS PubMed.
- N. J. Babu and A. Nangia, Cryst. Growth Des., 2011, 11, 2662–2679 CrossRef CAS.
- R. Ghadi, A. Ghuge, S. Ghumre, N. Waghmare and V. J. Kadam, Indo Am. J. Pharm. Res., 2014, 4(7), 3881–3892 Search PubMed.
- K. Raza, P. Kumar, S. Ratan, R. Malik and S. Arora, SOJ Pharm. Pharm. Sci., 2014, 1(2), 10 Search PubMed.
- Y. Matsuda, R. Akazawa, R. Teraoka and M. Otsuka, J. Pharm. Pharmacol., 1994, 46, 162–167 CrossRef CAS PubMed.
- R. J. Roberts, R. S. Payne and R. C. Rowe, Eur. J. Pharm. Sci., 2000, 9, 277–283 CrossRef CAS PubMed.
- P. Di Martino, A.-M. Guyot-Hermann, P. Conflant, M. Drache and J.-C. Guyot, Int. J. Pharm., 1996, 128, 1–8 CrossRef.
- G. Nichols and C. S. Frampton, J. Pharm. Sci., 1998, 87, 684–693 CrossRef CAS PubMed.
- T. Beyer, G. M. Day and S. L. Price, J. Am. Chem. Soc., 2001, 123, 5086–5094 CrossRef CAS PubMed.
- M. Otsuka, M. Onoe and Y. Matsuda, Pharm. Res., 1993, 10, 577–582 CrossRef CAS PubMed.
- R. Chadha, P. Arora, A. Saini and D. Singh Jain, J. Pharm. Pharm. Sci., 2012, 15(2), 234–251 CAS.
- N. Zencirci, U. J. Griesser, T. Gelbrich, D. C. Apperley and R. K. Harris, Mol. Pharmaceutics, 2013, 11, 338–350 CrossRef PubMed.
- N. Madusanka, M. D. Eddleston, M. Arhangelskis and W. Jones, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2014, 70, 72–80 CrossRef CAS PubMed.
-
U. J. Griesser, The Importance of Solvates, Wiley-VCH Verlag GmbH & Co. KGaA, 2006, pp. 211–233 Search PubMed.
- Mekinist – CHMP assessment report, https://www.ema.europa.eu/en/documents/assessment-report/mekinist-epar-public-assessment-report_en.pdf (access 17, July 2019).
- Forxiga – assessment report, https://www.ema.europa.eu/en/documents/assessment-report/forxiga-epar-public-assessment-report_en.pdf (access 17, July 2019).
- Prezista – Annex I, Summary of product characteristics, https://www.ema.europa.eu/en/documents/product-information/prezista-epar-product-information_en.pdf (access 17, July 2019).
- L. R. Chen, V. G. Jr. Young, D. Lechuga-Ballesteros and D. J. Grant, J. Pharm. Sci., 1999, 88(11), 1191–1200 CrossRef CAS PubMed.
- Sprycel – Annex I, Summary of product characterictics, http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/000709/WC500056998.pdf (access 17, July 2019).
- Keflex Capsules – https://www.accessdata.fda.gov/drugsatfda_docs/label/2006/050405s097lbl.pdf (access 17, July 2019).
- O. N. Kavanagh, D. M. Croker, G. M. Walker and M. J. Zaworotko, Drug Discovery Today, 2019, 24, 796–804 CrossRef CAS PubMed.
- M. B. Hickey, M. L. Peterson, L. A. Scoppettuolo, S. L. Morrisette, A. Vetter, H. Guzmán, J. F. Remenar, Z. Zhang, M. D. Tawa and S. Haley, Eur. J. Pharm. Biopharm., 2007, 67, 112–119 CrossRef CAS PubMed.
-
M. Zegarac, Pharmaceutically acceptable co crystalline forms of sildenafil, WO080362A1, 2007.
-
S. N. Devarakonda, K. Vyas, S. R. Bommareddy, P. R. Padi and B. Raghupathy, Aripiprazole co-crystals, WO/2007/092779, 2007.
- Tenofovir Disoproxil Hemi-Fumaric Acid Co-Crystal, 2007, www.faqs.org/patents/app/20090176983 (access 17, July 2019).
- A. Fini, G. Fazio, M.-J. F. Hervás, M. A. Holgado and A. M. Rabasco, Eur. J. Pharm. Sci., 1996, 4, 231–238 CrossRef CAS.
- D. Gupta, D. Bhatia, V. Dave, V. Sutariya and S. Varghese Gupta, Molecules, 2018, 23(7), 1719 CrossRef PubMed.
- P. Sanphui and A. Nangia, J. Chem. Sci., 2014, 126, 1249–1264 CrossRef CAS.
- G. S. Paulekuhn, J. B. Dressman and C. Saal, J. Med. Chem., 2007, 50, 6665–6672 CrossRef CAS PubMed.
- J. D. Dunitz and J. Bernstein, Acc. Chem. Res., 1995, 28, 193–200 CrossRef CAS.
- J. Bauer, S. Spanton, R. Henry, J. Quick, W. Dziki, W. Porter and J. Morris, Pharm. Res., 2001, 18, 859–866 CrossRef CAS PubMed.
- S. R. Chemburkar, J. Bauer, K. Deming, H. Spiwek, K. Patel, J. Morris, R. Henry, S. Spanton, W. Dziki, W. Porter, J. Quick, P. Bauer, J. Donaubauer, B. A. Narayanan, M. Soldani, D. Riley and K. McFarland, Org. Process Res. Dev., 2000, 4, 413–417 CrossRef CAS.
- C. H. Görbitz, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1997, 53, 736–739 CrossRef.
- P. Van Der Sluis and J. Kroon, J. Cryst. Growth, 1989, 97, 645–656 CrossRef CAS.
- A. M. Healy, Z. A. Worku, D. Kumar and A. M. Madi, Adv. Drug Delivery Rev., 2017, 117, 25–46 CrossRef CAS PubMed.
- S. Kumar and A. Nanda, Indian J. Pharm. Sci., 2017, 79(6), 858–871 Search PubMed.
- N. Schultheiss and A. Newman, Cryst. Growth Des., 2009, 9, 2950–2967 CrossRef CAS PubMed.
- R. Thakuria, A. Delori, W. Jones, M. P. Lipert, L. Roy and N. Rodríguez-Hornedo, Int. J. Pharm., 2013, 453, 101–125 CrossRef CAS PubMed.
- M. D. Eddleston, R. Thakuria, B. J. Aldous and W. Jones, J. Pharm. Sci., 2014, 103, 2859–2864 CrossRef CAS PubMed.
- R. Thakuria, M. Arhangelskis, M. D. Eddleston, E. H. H. Chow, K. K. Sarmah, B. J. Aldous, J. F. Krzyzaniak and W. Jones, Org. Process Res. Dev., 2019, 23, 845–851 CrossRef CAS.
- M. Gołdyn, D. Larowska, W. Nowak and E. Bartoszak-Adamska, CrystEngComm, 2019, 21, 5721–5732 RSC.
- D.-K. Bučar, R. F. Henry, G. G. Z. Zhang and L. R. MacGillivray, Cryst. Growth Des., 2014, 14, 5318–5328 CrossRef.
- D.-K. Bučar, R. F. Henry, X. Lou, R. W. Duerst, L. R. MacGillivray and G. G. Z. Zhang, Cryst. Growth Des., 2009, 9, 1932–1943 CrossRef.
- C. A. Guanawardana and C. B. Aakeröy, Chem. Commun., 2018, 54, 14047–14060 RSC.
- C. B. Aakeröy, A. M. Beatty and B. A. Helfrich, Angew. Chem., Int. Ed., 2001, 40, 3240–3242 CrossRef.
- B. R. Bhogala and A. Nangia, New J. Chem., 2008, 32, 800 RSC.
- E. Shefter, T. F. Brennan and P. Sackman, Chem. Pharm. Bull., 1971, 19, 746–752 CrossRef CAS.
- F. Fischer, M. Joester, K. Rademann and F. Emmerling, Chem. – Eur. J., 2015, 21, 14969–14974 CrossRef CAS PubMed.
- F. Fischer, G. Scholz, L. Batzdorf, M. Wilke and F. Emmerling, CrystEngComm, 2015, 17, 824–829 RSC.
- S. Karki, L. Fábián, T. Friščić and W. Jones, Org. Lett., 2007, 9, 3133–3136 CrossRef CAS PubMed.
- A. J. Cruz-Cabeza, S. Karki, L. Fábián, T. Friščić, G. M. Day and W. Jones, Chem. Commun., 2010, 46, 2224 RSC.
- N. Madusanka, M. D. Eddleston, M. Arhangelskis and W. Jones, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2014, 70, 72–80 CrossRef CAS PubMed.
- L. Vella-Zarb, D. Braga, A. Guy Orpen and U. Baisch, CrystEngComm, 2014, 16, 8147 RSC.
- H. D. Clarke, K. K. Arora, H. Bass, P. Kavuru, T. T. Ong, T. Pujari, L. Wojtas and M. J. Zaworotko, Cryst. Growth Des., 2010, 10, 2152–2167 CrossRef CAS.
- F. M. Amombo Noa and G. Mehlana, CrystEngComm, 2018, 20, 896–905 RSC.
- A. Jacobs and F. M. Amombo Noa, CrystEngComm, 2015, 17, 98–106 RSC.
- M. Habgood and S. L. Price, Cryst. Growth Des., 2010, 10, 3263–3272 CrossRef CAS.
- T. Rajbongshi, K. K. Sarmah, A. Sarkar, R. Ganduri, S. Cherukuvada, T. S. Thakur and R. Thakuria, Cryst. Growth Des., 2018, 18, 6640–6651 CrossRef CAS.
- K. K. Sarmah, T. Rajbongshi, S. Bhowmick and R. Thakuria, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2017, 73, 1007–1016 CrossRef CAS PubMed.
- R. Kaur, B. V. Lalithalakshmi and T. N. Guru Row, Cryst. Growth Des., 2014, 14, 2614–2620 CrossRef CAS.
- K. Ghosh, M. Datta, R. Fröhlich and N. C. Ganguly, J. Mol. Struct., 2005, 737, 201–206 CrossRef CAS.
- Y. Zhao, S. Jin, Z. Tao, Y. Lin, L. Wang, D. Wang, J. Guo and M. Guo, J. Chem. Crystallogr., 2016, 46, 188–202 CrossRef CAS.
- K. K. Sarmah, K. Boro, M. Arhangelskis and R. Thakuria, CrystEngComm, 2017, 19, 826–833 RSC.
- L. S. Reddy, S. J. Bethune, J. W. Kampf and N. Rodríguez-Hornedo, Cryst. Growth Des., 2009, 9, 378–385 CrossRef CAS.
-
Agilent, CrysAlis PRO, Agilent Technologies Ltd, Yarnton, Oxfordshire, England, 2014 Search PubMed.
-
Oxford Diffraction, CrysAlis RED, Oxford Diffraction Ltd, Abingdon, Oxfordshire, England, 2006 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- Kdif Software, Karel Knížek, https://www.fzu.cz/~knizek/kalvados/download.html (access on 29, July 2019).
- C. F. Macrae, P. R. Edgington, P. McCabe, E. Pidcock, G. P. Shields, R. Taylor, M. Towler and J. van de Streek, J. Appl. Crystallogr., 2006, 39, 453–457 CrossRef CAS.
- FOODB, http://foodb.ca/compounds/FDB000455 (access on 29, July 2019).
- A. J. Cruz-Cabeza, CrystEngComm, 2012, 14, 6362 RSC.
- M. K. Corpinot and D.-K. Bučar, Cryst. Growth Des., 2018, 19, 1426–1453 CrossRef.
- M. C. Etter, Acc. Chem. Res., 1990, 23, 120–126 CrossRef CAS.
- B. Sarma, P. Sanphui and A. Nangia, Cryst. Growth Des., 2010, 10, 2388–2399 CrossRef CAS.
- ECMBD, http://ecmdb.ca/compounds/ECMDB24184 (access on 29, July 2019).
- ChemicalBook, https://www.chemicalbook.com/ChemicalProductProperty_EN_CB4467889.htm (access on 29, July 2019).
-
S. H. Yalkowsky and H. Yan, Handbook of aqueous solubility data, CRC Press, 2003 Search PubMed.
- TGSC Information System, http://www.thegoodscentscompany.com/data/rw1166771.html (access on 29, July 2019).
- DrugBank, https://www.drugbank.ca/drugs/DB03946 (access on 29, July 2019).
- FOODB, http://foodb.ca/compounds/FDB000848 (access on 29, July 2019).
- X. Liao, M. Gautam, A. Grill and H. J. Zhu, J. Pharm. Sci., 2010, 99, 246–254 CrossRef CAS PubMed.
- C. R. Groom, I. J. Bruno, M. P. Lightfoot and S. C. Ward, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2016, 72, 171–179 CrossRef CAS PubMed.
- Z.-L. Wang and L.-H. Wei, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2007, 63, o1681–o1682 CrossRef CAS.
- M. J. Mnguni, J. P. Michael and A. Lemmerer, Acta Crystallogr., Sect. C: Struct. Chem., 2018, 74, 715–720 CrossRef CAS PubMed.
- S. Aitipamula, P. S. Chow and R. B. H. Tan, CrystEngComm, 2012, 14, 2381 RSC.
- G. R. Desiraju, J. Chem. Soc., Chem. Commun., 1991, 426 RSC.
- T. R. Shattock, K. K. Arora, P. Vishweshwar and M. J. Zaworotko, Cryst. Growth Des., 2008, 8, 4533–4545 CrossRef CAS.
- S. Cherukuvada and T. N. Guru Row, Cryst. Growth Des., 2014, 14, 4187–4198 CrossRef CAS.
- K. D. Prasad, S. Cherukuvada, R. Ganduri, L. D. Stephen, S. Perumalla and T. N. Guru Row, Cryst. Growth Des., 2015, 15, 858–866 CrossRef CAS.
- E. Lu, N. Rodríguez-Hornedo and R. Suryanarayanan, CrystEngComm, 2008, 10, 665 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Stacking interaction geometry in described TBR cocrystals (Table S1); steady state absorption calibration curves of TBR·23DHBA·H2O, TBR·24DHBA, TBR·25DHBA, (TBR-H)+·(26DHBA)−·(H2O), TBR·34DHBA and TBR·35DHBA (Fig. S1). CCDC 1938121, 1938122, 1938144, 1938146, 1938148 and 1938149. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9ce01195a |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *,
Daria
Larowska
*,
Daria
Larowska