
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Sequential halochromic/mechanochromic luminescence of pyridyl-substituted solid-state emissive dyes: thermally controlled stepwise recovery of the original emission color†
Suguru
Ito
 *,
Chika
Nishimoto
and
Sayaka
Nagai
*,
Chika
Nishimoto
and
Sayaka
Nagai
Department of Advanced Materials Chemistry, Graduate School of Engineering, Yokohama National University, 79-5 Tokiwadai, Hodogaya-ku, Yokohama 240-8501, Japan. E-mail: suguru-ito@ynu.ac.jp
First published on 19th August 2019
Abstract
Stimuli-responsive organic dyes that switch the color of their solid-state emission upon exposure to external stimuli represent an important class of materials due to their potential applications in various areas of sensing technology. Although an increasing number of multi-stimuli-responsive organic dyes have been reported in recent years, only a few solid-state fluorophores are known to respond to two kinds of stimuli in a stepwise and reversible manner. Herein, we report the sequential halochromic (acidochromic)/mechanochromic luminescence behavior of a pyridyl-substituted solid-state fluorophore. The solid-state emission color of this pyridyl-substituted dye was systematically shifted in the bathochromic direction by forming cocrystals with several benzoic acid derivatives that exhibit different pKa values. Upon grinding these cocrystals with a spatula, further bathochromic shifts of their maximum emission wavelengths were observed upon amorphization. Conversely, the emission color and crystallinity can be recovered upon heating the ground samples of the cocrystals to their cold-crystallization transition temperature (Tc ∼ 100 °C). Heating the ground cocrystals further (185–260 °C) removes the benzoic acid derivatives, which restores the original emission color of the pyridyl-substituted dye. In other words, we have developed a new system that exhibits a two-step emission color recovery in response to temperature by combining halochromic and mechanochromic luminescence in series.
Introduction
Solid-state luminescent materials that respond to external stimuli are of great interest due to their potential applications in fluorescence sensors and optical switches.1,2 Mechanical stimuli such as grinding, crushing, and shearing have recently received much attention, and a growing number of organic dyes that exhibit mechanochromic luminescence (MCL) have emerged in the past decade.3–8 The mechanically switched emission color of MCL dyes reverts to the original color upon exposure to a second external stimulus such as heating or exposure to solvents. As most MCL dyes switch their emission color between two colors,4 other external stimuli are required to achieve multi-color switching of their solid-state emission. Several MCL dyes exhibit multi-stimuli-responsive properties, whereby the solid-state emission color of the initial state switches to different colors in response to other external stimuli.5,6 Among these, some MCL dyes with basic functional groups have been shown to exhibit halochromic (acidochromic) luminescence (HCL) that can change their solid-state emission color by forming acid–base complexes with external acids.5 However, little is known about the mechano-responsive behavior of such acid–base complexes.7 In other words, typical dyes that display HCL and MCL behavior independently respond to acid and mechanical stimuli (Fig. 1a).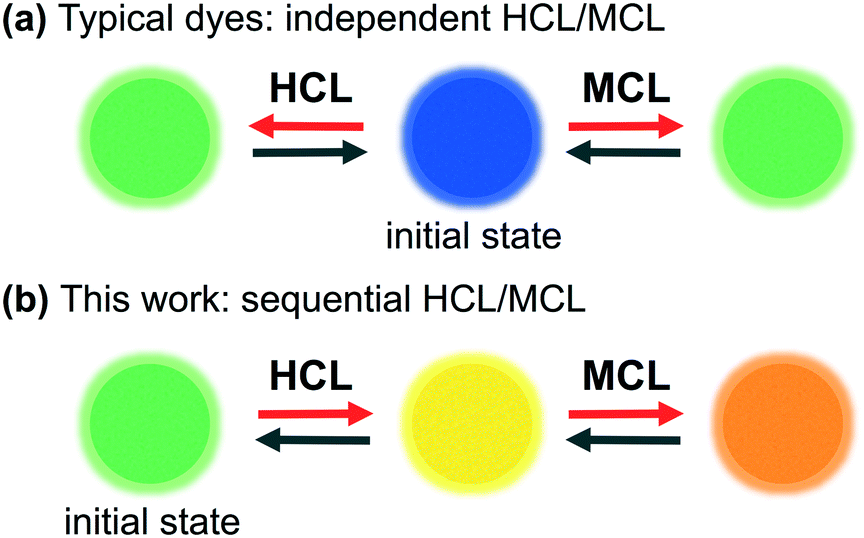 | ||
| Fig. 1 Schematic illustration of the multi-stimuli-responsive behavior of organic dyes. (a) Typical system: independent HCL/MCL; (b) this work: sequential HCL/MCL. | ||
During our studies on the development of MCL dyes composed of electron-donating and -accepting heteroaromatic rings,8 we recently reported phenanthroimidazolylbenzothiadiazoles as a new class of MCL dyes that exhibit versatile MCL properties.8a,9 The phenanthroimidazole ring10 is constructed from 9,10-phenanthrenequinone, ammonium acetate, aniline, and 4-bromo-7-formylbenzothiadiazole. Various substituted benzene rings can be easily introduced by replacing the bromo group of the resulting phenanthroimidazolylbenzothiadiazole. Herein, we report the multi-stimuli-responsive properties of pyridyl-substituted phenanthroimidazolylbenzothiadiazole derivative 1. The emission properties of crystalline 1 are sensitive to mechanical stimuli as well as to complexation with benzoic acid derivatives 2. Notably, we accomplished a stepwise stimuli-responsive change of the emission color, whereby the solid-state emission wavelength of 1 shifts bathochromically upon sequential exposure to acids and mechanical stimuli. Furthermore, the mechanically changed state exhibits a two-step hypsochromic shift of the emission color in response to heating (Fig. 1b).
Results and discussion
Pyridyl-substituted phenanthroimidazolylbenzothiadiazole 1 was synthesized from previously reported bromide 3.8a A Suzuki–Miyaura coupling reaction between 3 and pyridin-4-ylboronic acid in the presence of Pd(PPh3)4 (10 mol%) in DMF/aqueous K2CO3 (1.4 M) at 150 °C (bath temp.) afforded 1 in 88% yield (Scheme 1). The following experiments were carried out using crystalline 1, which was prepared from vapor diffusion of hexane into a chloroform/DMF solution of 1 (Fig. S1†).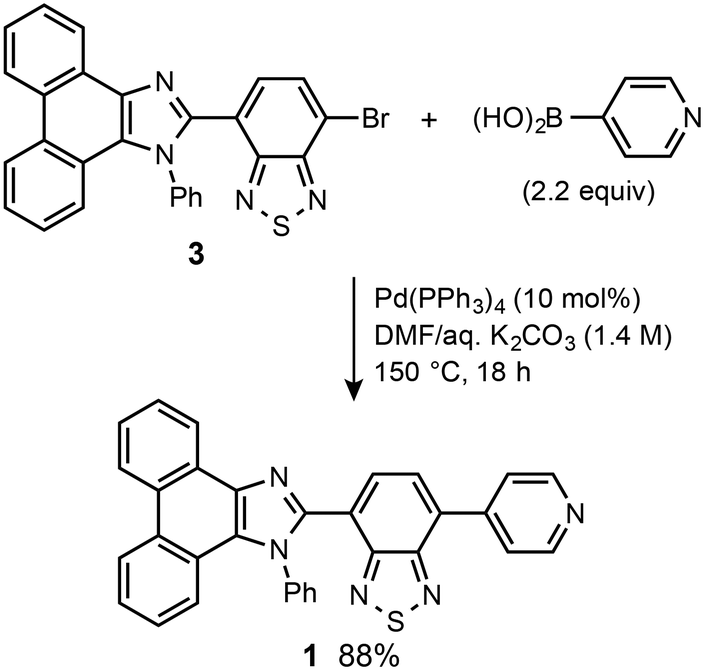 | ||
| Scheme 1 Synthesis of pyridyl-substituted phenanthroimidazolylbenzothiadiazole 1. | ||
Crystalline samples of 1 exhibit a green emission (λem = 521 nm) with a good fluorescence quantum yield (ΦF = 0.34). Upon strong grinding with a spatula, the emission color of crystalline 1 shifted bathochromically to orange (λem = 593 nm; ΦF = 0.35) (Fig. 2a). Powder X-ray diffraction (PXRD) analyses of crystalline and ground 1 showed that the intensity of the diffraction peaks of crystalline 1 significantly decreases after grinding, which indicates the amorphization of the crystals (Fig. 2b). Moreover, differential scanning calorimetry (DSC) measurements of crystalline 1 exhibited one endothermic peak that corresponds to the melting point (Tm = 325 °C), whereas for ground 1, an exothermic cold-crystallization transition peak (Tc = 124 °C) and Tm were observed (Fig. 2c). Upon heating the ground sample of 1 to its Tc, the orange emission color reverted to the original green emission color (Fig. 2a). Therefore, 1 exhibits MCL between green and orange, whereby the change of the emission color should be attributed to the crystalline-to-amorphous phase transition.
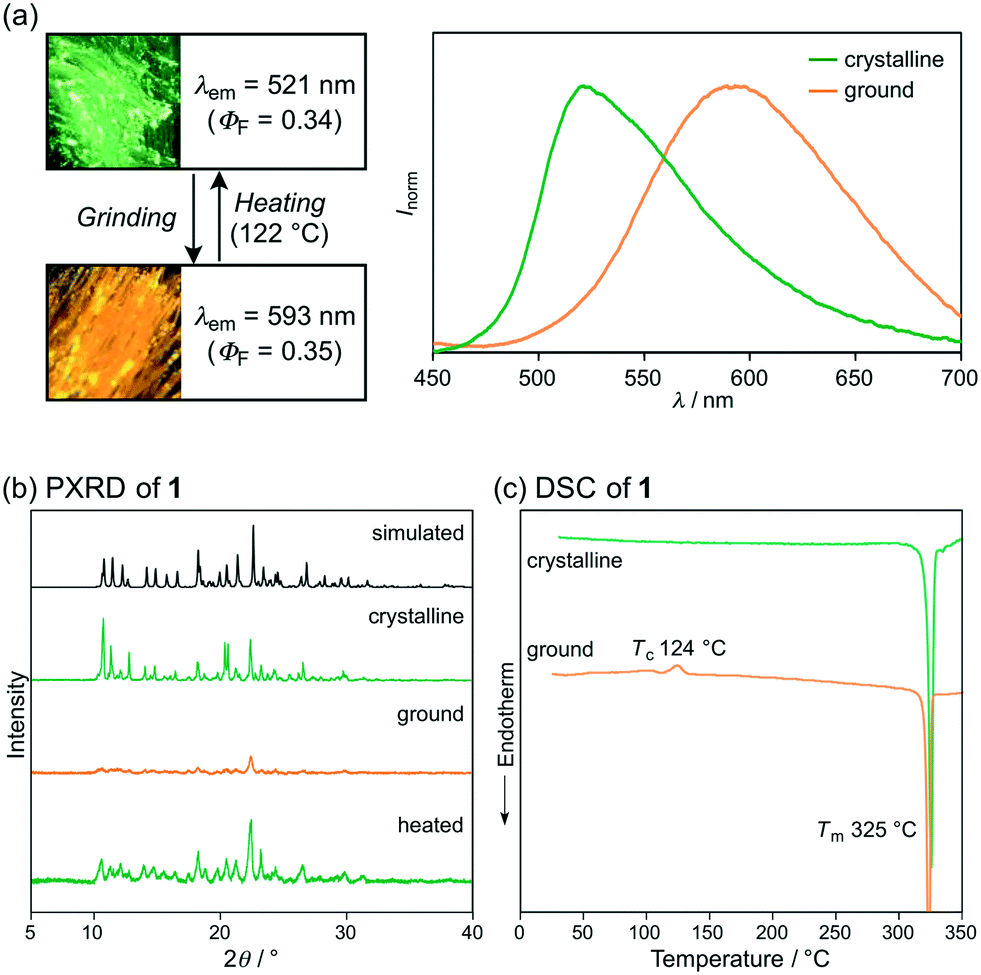 | ||
| Fig. 2 (a) Photographs and fluorescence spectra, (b) PXRD patterns, and (c) DSC scans for the MCL of 1. Tc and Tm values are noted near their corresponding peaks. | ||
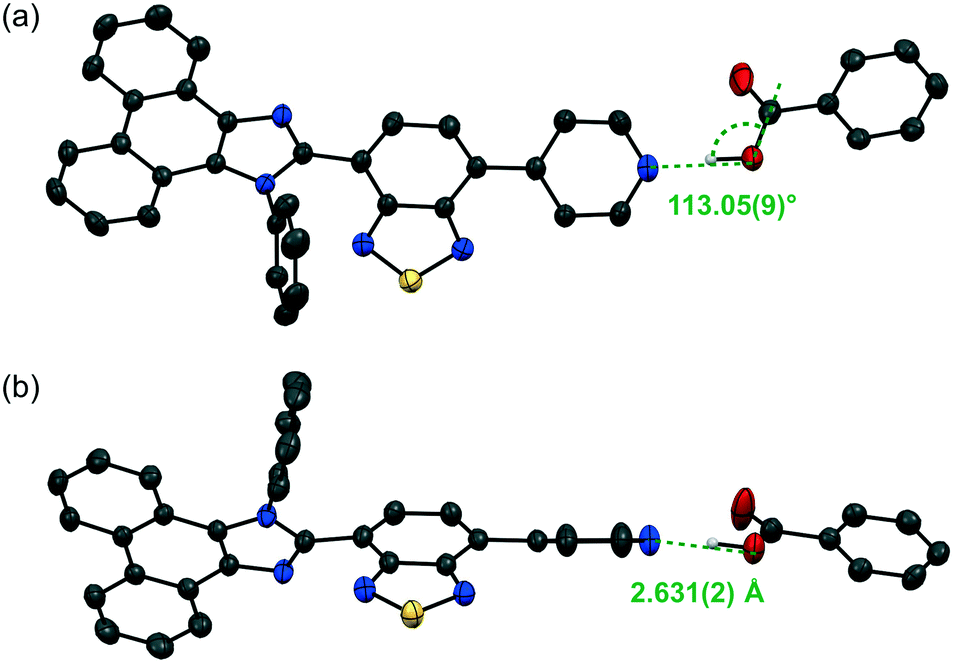
With pyridyl-substituted MCL-active dye 1 in hand, the acid-responsive properties of 1 were examined by preparing acid–base complexes with benzoic acid derivatives 2a–d (RC6H4CO2H; Table 1).11 Cocrystals of 1 and benzoic acid (2a: R = H) were obtained from vapor diffusion of hexane into a chloroform solution of a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar mixture of 1 and 2a. The maximum emission wavelength of cocrystal 1·2a shifted bathochromically to 551 nm (ΦF = 0.19) compared to that of crystalline 1 (Fig. 3a). The single-crystal X-ray diffraction analysis of 1·2a revealed that the cocrystal (P21/n) is composed of a 1:1 molar mixture of 1 and 2a, which forms O–H⋯N hydrogen bonds between the pyridyl groups of 1 and the carboxy groups of 2a (N⋯O distance = 2.631 Å; Fig. 4 and S2†). In a similar fashion, other cocrystals (P21/n) were prepared using 2b–d, which contain electron-withdrawing substituents (2b: R = CF3; 2c: R = CN; 2d: R = NO2) on the 4-position of benzoic acid (Fig. S3–S5†); the resulting cocrystals exhibit yellow to orange emission with good fluorescence quantum yields (λem = 562–593 nm; ΦF = 0.23–0.36; Fig. 3b–d). Interestingly, the maximum emission wavelength of these cocrystals shifts in the bathochromic direction (1·2a: 551 nm; 1·2b: 562 nm; 1·2c: 590 nm; 1·2d: 593 nm) with decreasing pKa value of the benzoic acid derivative (2a: 4.19; 2b: 3.66; 2c: 3.55; 2d: 3.44).12 On the other hand, the solid-state absorption spectra of 1·2a–d, obtained by measuring the diffuse reflectance spectra, were observed in almost the same region as that of crystalline 1, although the maximum absorption wavelength of 1·2a–d slightly shifted in the bathochromic direction upon increasing the acidity of 2a–d (Fig. S6 and S7†).
:1 molar mixture of 1 and 2a. The maximum emission wavelength of cocrystal 1·2a shifted bathochromically to 551 nm (ΦF = 0.19) compared to that of crystalline 1 (Fig. 3a). The single-crystal X-ray diffraction analysis of 1·2a revealed that the cocrystal (P21/n) is composed of a 1:1 molar mixture of 1 and 2a, which forms O–H⋯N hydrogen bonds between the pyridyl groups of 1 and the carboxy groups of 2a (N⋯O distance = 2.631 Å; Fig. 4 and S2†). In a similar fashion, other cocrystals (P21/n) were prepared using 2b–d, which contain electron-withdrawing substituents (2b: R = CF3; 2c: R = CN; 2d: R = NO2) on the 4-position of benzoic acid (Fig. S3–S5†); the resulting cocrystals exhibit yellow to orange emission with good fluorescence quantum yields (λem = 562–593 nm; ΦF = 0.23–0.36; Fig. 3b–d). Interestingly, the maximum emission wavelength of these cocrystals shifts in the bathochromic direction (1·2a: 551 nm; 1·2b: 562 nm; 1·2c: 590 nm; 1·2d: 593 nm) with decreasing pKa value of the benzoic acid derivative (2a: 4.19; 2b: 3.66; 2c: 3.55; 2d: 3.44).12 On the other hand, the solid-state absorption spectra of 1·2a–d, obtained by measuring the diffuse reflectance spectra, were observed in almost the same region as that of crystalline 1, although the maximum absorption wavelength of 1·2a–d slightly shifted in the bathochromic direction upon increasing the acidity of 2a–d (Fig. S6 and S7†).
| 2 | R | pKaa | Crystalline complexb | Space groupd | N⋯Oe (Å) | Calcd. λabsf (nm) | λ abs (nm) | T c (°C) | Ground complexb | |||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| (in H2O) | λ em (nm) | Φ F | 1(·2) | 1·2 | 1·2 | λ em (nm) | Φ F | |||||
| a pKa values of 2a–d in H2O. b Maximum emission bands (λem) and absolute quantum yields (ΦF) of 1·2a–d in the crystalline powdered form and in the ground amorphous form. c Absolute quantum yields measured using an integrating sphere. d Space group of cocrystals 1·2a–d. e Distance between the hydrogen-bond forming nitrogen atom of the pyridyl group in 1 and the oxygen atom of the carboxy group in 2a–d. f Maximum absorption wavelength of 1(·2) and 1·2a–d, calculated at the CAM-B3LYP/6-31G(d) level of theory. g Maximum absorption wavelength of crystalline 1·2a–d. The values for the ground samples are shown in the parentheses (Fig. S6 and S7). h Cold-crystallization transition temperature of 1·2a–d. | ||||||||||||
| 2a | H | 4.19 | 551 | 0.19 | P21/n | 2.631 | 366.65 | 379.96 | 415 (425) | 86 | 594 | 0.34 |
| 2b | CF3 | 3.66 | 562 | 0.23 | P21/n | 2.622 | 362.65 | 382.77 | 417 (429) | 90 | 610 | 0.30 |
| 2c | CN | 3.55 | 590 | 0.36 | P21/n | 2.594 | 371.73 | 386.76 | 419 (433) | 87 | 616 | 0.40 |
| 2d | NO2 | 3.44 | 593 | 0.33 | P21/n | 2.595 | 368.35 | 385.37 | 422 (437) | 103 | 627 | 0.22 |
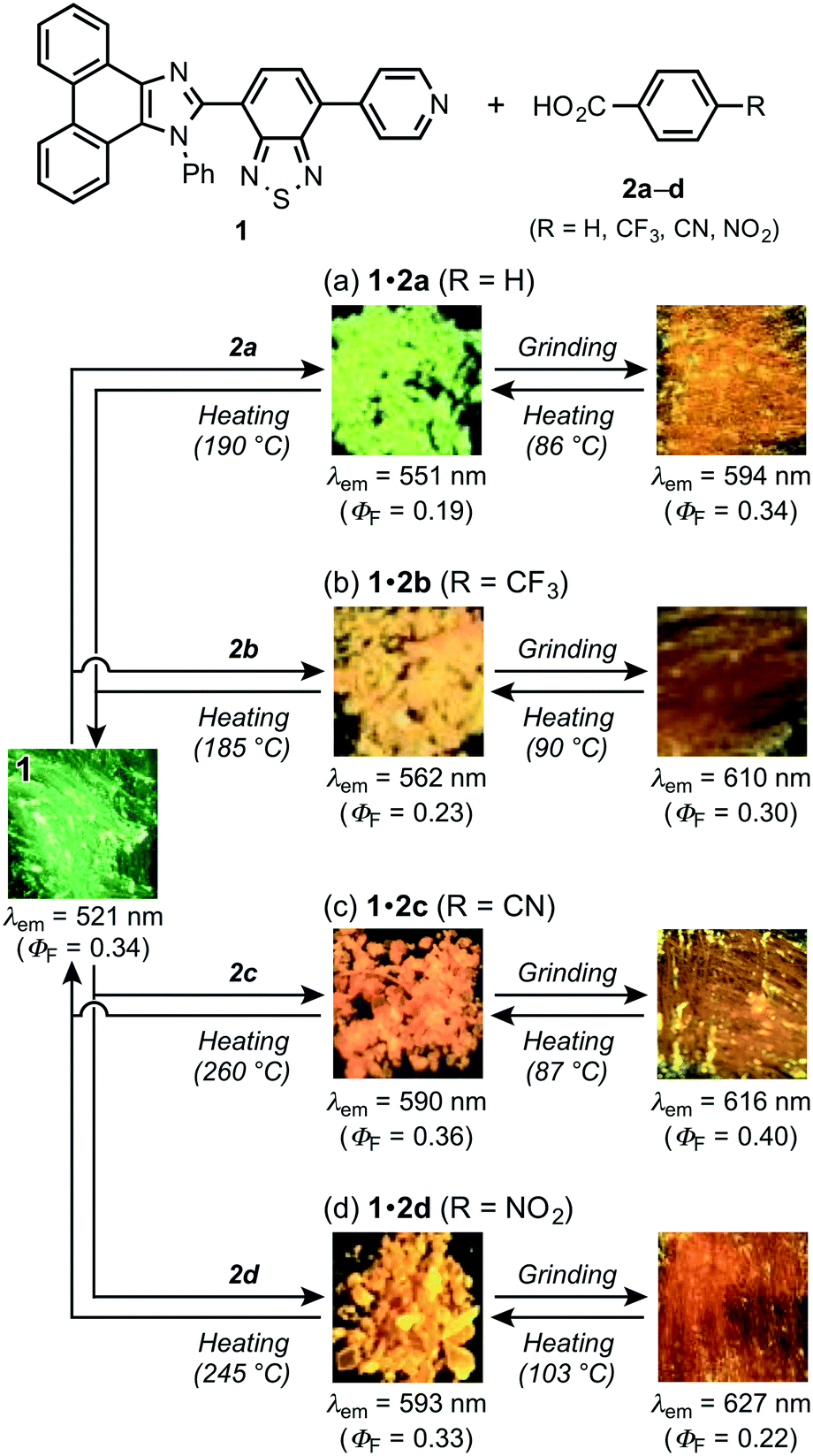 | ||
| Fig. 3 Structures and photographs for the sequential HCL/MCL of 1·2a–d. MCL of 1·2a (a), 1·2b (b), 1·2c (c), and 1·2d (d). | ||
 | ||
| Fig. 4 (a) Front view and (b) side view of the molecular structure of 1·2a with thermal ellipsoids at 50% probability. All hydrogen atoms, except for that of the carboxy group, are omitted for clarity. Color code: gray = C; white = H; red = O; blue = N; yellow = S. | ||
The molecular structures obtained by single-crystal X-ray diffraction analyses of the cocrystals showed that the distance between the hydrogen-bond-forming pyridyl group of 1 and the carboxy group of 2 decreases with increasing acidity of the benzoic acid derivative 2 [N⋯O distances (Å); 1·2a: 2.631; 1·2b: 2.622; 1·2c: 2.594; 1·2d: 2.595]. It has been shown previously that the absorption and emission wavelengths of phenanthroimidazolylbenzothiadiazole derivatives depend on their conformational structure.8a Based on the single-crystal X-ray diffraction structures, the absorption wavelengths of 1 in 1·2a–d [denoted as 1(·2a–d)] and discrete hydrogen-bond-forming complexes 1·2a–d were calculated using time-dependent density functional theory (TD-DFT) at the CAM-B3LYP/6-31G(d) level of theory (Tables 1 and S1, Fig. S8 and S9†). The calculated absorption wavelength values reflect the conformational structures of 1(·2a–d), whereby 1(·2c), which exhibits the most planar structure, shows the largest value (calcd. λabs = 371.73 nm). However, these values cannot explain the order of the experimentally observed λabs values for 1·2a–d (1·2a: 415 nm; 1·2b: 417 nm; 1·2c: 419 nm; 1·2d: 422 nm), as 1(·2a) exhibits a relatively large value (calcd. λabs = 366.65 nm). On the other hand, significant bathochromic shifts were observed for the calculated λabs values of the acid–base complexes (1·2a: 379.96 nm; 1·2b: 382.77 nm; 1·2c: 386.76 nm; 1·2d: 385.37 nm) compared to those of 1 (362.65–371.73 nm) in these complexes. The order of calculated λabs values for 1·2a–d is similar to that of experimental values. As the calculations were performed on discrete complexes, the difference between the order of the theoretical and experimental values could potentially be explained by considering the electronic intermolecular interaction of the complexes, which would mean that the electronic rather than the steric effects should have a significant influence on the optical properties of 1·2a–d.
It should be noted here that cocrystals were not obtained from mixtures of 1 and 4-(methoxycarbonyl)benzoic or 3,5-dinitrobenzoic acid, regardless of their higher acidity relative to that of 2a. From these mixtures, both components precipitated as independent crystals. Accordingly, the steric bulk of the substituents on the benzene ring of the benzoic acid derivative should also be important for the formation of acid–base cocrystals of the P21/n space group.
Subsequently, we examined the mechanical-stimuli-responsive properties of cocrystal 1·2a (Fig. 3a). Upon grinding the crystalline samples of 1·2a with a spatula, the maximum emission wavelength shifted bathochromically to 594 nm (ΦF = 0.34), which is almost identical to that of ground 1 (λem = 593 nm). A significantly decreased intensity of the diffraction patterns of cocrystal 1·2a was observed by the PXRD analysis of the ground state of 1·2a, indicating that the mechanism of the mechano-responsive change of the emission color of 1·2a is based on typical crystal-to-amorphous transitions (Fig. 5a). The DSC thermogram of ground 1·2a showed an exothermic peak at 86 °C, which corresponds to Tc (Fig. 5b). Upon heating the ground sample of 1·2a to its Tc, the emission color and PXRD intensities were reverted to those of the original cocrystal 1·2a. In other words, cocrystal 1·2a exhibits MCL between yellow (λem = 551 nm) and orange (λem = 594 nm). We also examined complex 1·2a prepared by grinding 1 and 2a in the solid state. Upon grinding the crystalline samples of 1 and 2a, the mixture changed to a sticky paste-like state that exhibited yellow emission (λem = 562 nm). Although this yellow-emissive state was prepared by grinding, the emission color was changed to orange (λem = 590 nm) upon strong grinding with a spatula. In contrast to the ground state of cocrystal 1·2a, this orange-emissive state spontaneously reverted to the yellow-emissive state at room temperature within 5 min.
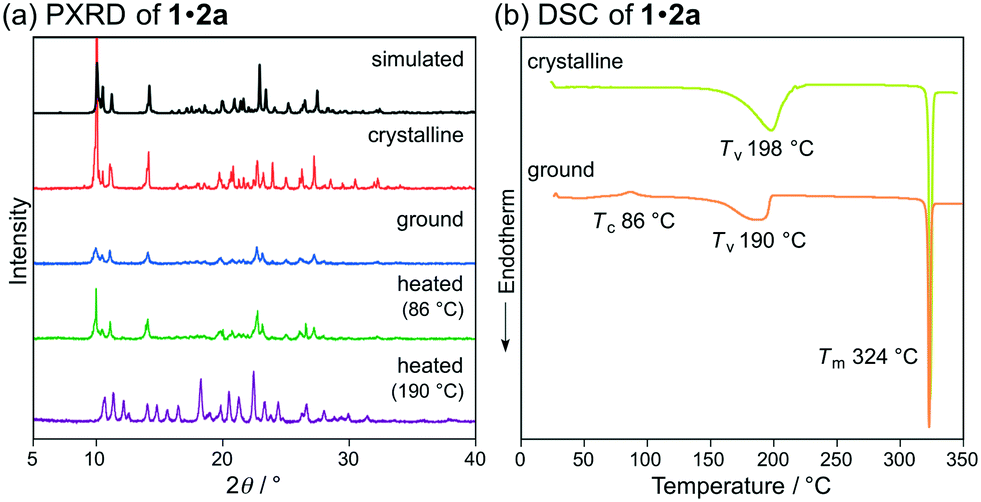 | ||
| Fig. 5 (a) PXRD patterns and (b) DSC scans for the MCL of 1·2a. Tc, Tv (vaporization temperature), and Tm values are noted near their corresponding peaks. | ||
Notably, two endothermic peaks were observed in the DSC thermogram of cocrystal 1·2a at 190 °C and 324 °C. When cocrystal 1·2a was heated to 190 °C, the emission color reverted to green that is characteristic of 1. The absence of 2a was confirmed by the 1H NMR spectrum of the green-emissive state after heating to 190 °C (Fig. S10†). This state also exhibited almost the same PXRD patterns as those of crystalline 1 (Fig. 2b and 5a), and the latter peak of the DSC thermogram (324 °C) corresponds to the Tm of 1 (Fig. 2c and 5b). In other words, 1 shows HCL between green (λem = 521 nm) and yellow (λem = 551 nm) in response to the addition and removal of 2a.
Similarly, significant bathochromic shifts of the maximum emission wavelengths were observed for cocrystals 1·2b–d upon grinding (2b: λem = 610 nm, ΦF = 0.30; 2c: λem = 616 nm, ΦF = 0.40; 2d: λem = 627 nm, ΦF = 0.22; Fig. 3b–d). In all cases, the intensities of the PXRD patterns decreased, which indicated the mechano-responsive amorphization of the cocrystals (Fig. S14–S16†). The maximum emission wavelengths of the ground samples were bathochromically shifted with increasing electron-withdrawing nature of the benzoic acid derivatives, which is probably due to the stabilization of the excited state of amorphous 1 by intermolecular interactions of the electron-deficient benzene rings. The original emission colors and PXRD patterns of the cocrystals were restored by heating the ground samples to their Tc (1·2b: 90 °C; 1·2c: 87 °C; 1·2d: 103 °C). On the other hand, the DSC thermograms of 1·2b–d exhibited endothermic peaks that correspond to the vaporization of 2b–d (1·2b: 185 °C; 1·2c: 260 °C; 1·2d: 245 °C) (Fig. S17–S19†). The recovery of the emission color, PXRD pattern, and 1H NMR signals (Fig. S11–S13†) of 1 was also observed upon heating 1·2b–d to the corresponding temperatures. Accordingly, all cocrystals 1·2a–d exhibited mechano- and thermo-responsive properties.
In order to confirm the effect of the pyridyl group, phenanthroimidazolylbenzothiadiazole 4,8a which bears a phenyl group instead of a pyridyl group, was treated with benzoic acid derivatives 2a–d (Fig. 6). In these cases, single crystals were not obtained from vapor diffusion of hexane into chloroform solutions of 1:1 molar mixtures of 4 and 2a–d. When CH2Cl2 solutions of 4 and 2a–d were evaporated under reduced pressure, the resulting mixtures of 4/2a–d exhibited yellow emission in the same region, irrespective of the acidity of the benzoic acid derivatives (4/2a: 564 nm; 4/2b: 570 nm; 4/2c: 567 nm; 4/2d: 564 nm). Notably, the emission color of these mixtures remained virtually unchanged after grinding with a spatula. The PXRD analyses of the yellow-emissive mixtures indicated that 4 is present in the amorphous form in these mixtures (Fig. S20–S23†). As the emission color of crystalline 4 changes from green to yellow by partial amorphization,8a the origin of the yellow emission from the mixture of 4 and 2a–d should be ascribed to the emission from amorphous 4. Moreover, the non-MCL properties of 4/2a–d should be rationalized in terms of the absence of crystal-to-amorphous transitions of 4 in 4/2a–d. These results indicate that the pyridyl group should play an important role in the sequential HCL/MCL of 1 by forming cocrystals based on hydrogen bonding with 2a–d.
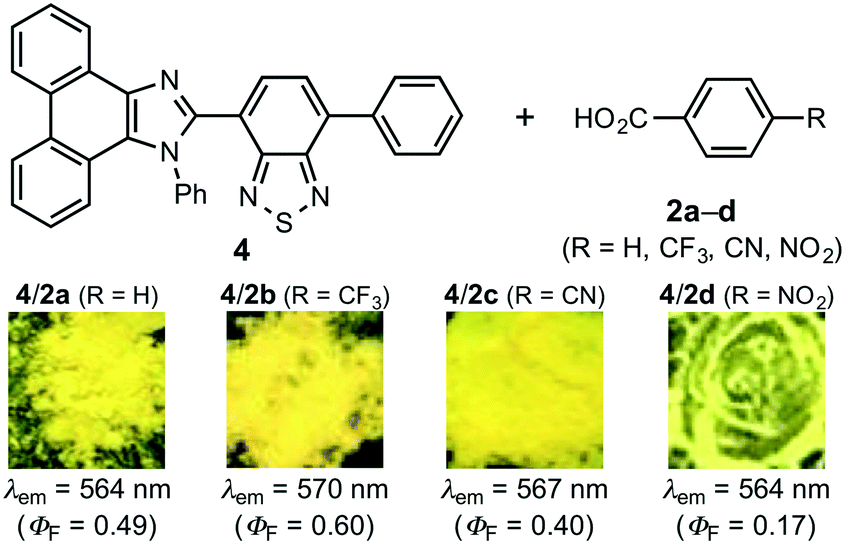 | ||
| Fig. 6 Photographs of the solid-state emission of mixtures of 4 and 2a–d. | ||
Conclusions
In summary, we have developed a new pyridyl-substituted mechanochromic luminescence (MCL) dye 1 that exhibits sequential halochromic luminescence (HCL)/MCL in response to treatment with benzoic acid derivatives 2a–d and mechanical stimuli. The solid-state emission color of 1 can be switched reversibly between two colors by addition and removal of acids 2a–d. Bathochromic shifts of the emission wavelength for cocrystals 1·2a–d relative to that of 1 can be attributed to the formation of hydrogen bonds between the pyridyl group of 1 and the carboxy group of 2a–d, whereby the magnitude of the shift increases with decreasing pKa value of 2a–d. Further bathochromic shifts were observed for cocrystals 1·2a–d upon grinding, and the ground amorphous state reverted to the crystalline 1·2a–d state upon heating the ground samples to their Tc. It should be noted that the two-step thermo-responsive change of the emission color can be realized from the ground samples of 1·2a–d by heating them to their Tc (∼100 °C) followed by heating to higher temperatures (>180 °C), which is required for the removal of 2a–d. The development of such a system, which combines HCL and MCL in series, should provide access to a variety of practical applications of MCL dyes.Experimental
General
All air-sensitive experiments were carried out under an argon atmosphere unless otherwise noted. IR spectra were recorded on a Nicolet iS10 FT-IR spectrometer. 1H and 13C NMR spectra were recorded on a Bruker DRX-500 spectrometer using tetramethylsilane as an internal standard. Fluorescence and UV-vis absorption spectra were measured on a JASCO FP-8300 fluorescence spectrometer. The solid-state absorption spectra were obtained by measuring diffuse reflectance spectra using an FPA-810 powder sample cell block. The absolute fluorescence quantum yields were determined using a 100 mm ϕ integrating sphere JASCO ILF-835. A miniature fiber-optic spectrometer (FLAME-S-XR1-ES, Ocean Optics) was used for the measurements of mechanochromic luminescence. PXRD measurements were performed on a Rigaku SmartLab system using CuKα radiation. The melting point was determined on a Stuart melting point apparatus SMP3 and was uncorrected. DSC data were recorded on a Shimadzu DSC-60 plus (heating rate: 10 °C min−1). The high-resolution electrospray ionization (HRMS-ESI) mass spectrum was recorded on a Hitachi Nano Frontier LD spectrometer. Silica gel 60 N (spherical, neutral, 63–210 mm) was used for column chromatography. 4-Bromo-7-(1-phenyl-1H-phenanthro[9,10-d]imidazol-2-yl)benzo[c][1,2,5]thiadiazole (3) was synthesized according to a literature procedure.8a Other reagents and solvents were commercially available and were used as received.Synthesis of 4-(1-phenyl-1H-phenanthro[9,10-d]imidazol-2-yl)-7-(pyridin-4-yl)benzo[c][1,2,5]thiadiazole (1)
A mixture of pyridin-4-ylboronic acid (22.0 mg, 0.29 mmol) and bromide 3 (67.8 mg, 0.13 mmol) in a mixture of DMF (4.0 mL) and an aqueous solution of K2CO3 (0.4 mL, 1.4 M) was degassed under ultrasonication. Pd(PPh3)4 (15.1 mg, 0.013 mmol) was added to the mixture, which was further degassed under ultrasonication. After the mixture was stirred at 150 °C (bath temp.) for 18 h, water and dichloromethane were added to the mixture. The organic layer was separated, and the aqueous layer was extracted three times with dichloromethane. The combined organic layer was washed with water and brine, dried over anhydrous Na2SO4, and filtered. After removal of the solvent under reduced pressure, the crude product was purified by silica-gel column chromatography (ethyl acetate/hexane = 10:1) to give 4-(1-phenyl-1H-phenanthro[9,10-d]imidazol-2-yl)-7-(pyridin-4-yl)benzo[c][1,2,5]thiadiazole (1: 59.4 mg, 88%) as a yellow solid.
4-(1-phenyl-1H-phenanthro[9,10-d]imidazol-2-yl)-7-(pyridin-4-yl)benzo[c][1,2,5]thiadiazole (1)
 Yellow solid; M.p. 319.8–321.9 °C; IR (KBr): νmax 1596, 1542, 1517, 1497, 1473, 1454, 1414, 1389, 1352, 1335, 1324, 1238, 1169, 1096, 1069, 1035, 994, 930, 885, 848, 825, 794, 764, 730, 721, 704, 686, 666, 614 cm−1; 1H NMR (500 MHz, CDCl3): δ (ppm) 8.86 (d, J = 7.8 Hz, 1H, Hd), 8.80 (d, J = 8.5 Hz, 1H, Hh), 8.78–7.77 (m, 2H, Hy2), 8.73 (d, J = 7.8 Hz, 1H, Ha), 7.88–7.87 (m, 2H, Hy1), 7.84 (d, J = 7.3 Hz, 1H, Hs), 7.77 (d, J = 7.3 Hz, 1H, Ht), 7.74 (t, J = 7.8 Hz, 1H, Hc), 7.67 (t, J = 7.8 Hz, 1H, Hb), 7.57–7.54 (m, 3H, Hi, Hp1), 7.45–7.40 (m, 3H, Hp2, Hp3), 7.31–7.27 (m, 2H, Hj, Hk); 13C NMR (126 MHz, CDCl3): δ (ppm) 154.3 (Cw), 152.7 (Cv), 150.2 (Cy2), 147.4 (Cq), 144.1 (Cx), 137.9 (Co, Cm), 132.3 (Cu), 132.0 (Cs), 129.64 (Cg), 129.59 (Cp3), 129.56 (Cp2), 128.9 (Cp1), 128.4 (Cf), 128.3 (Cn), 127.8 (Ct), 127.4 (Cc), 127.2 (Ce), 126.4 (Cj), 125.8 (Cb), 125.3 (Ci), 125.0 (Cr), 124.2 (Ch), 123.6 (Cy1), 123.1 (Ca), 122.9 (Cd, Cl), 121.1 (Ck); HRMS-ESI (m/z): [M + H]+ calcd. for C32H20N5S, 506.1434; found, 506.1448. Crystal data for 1 (CCDC 1935319): C32H19N5S, M = 505.60, monoclinic, a = 9.71088(5) Å, b = 16.39216(9) Å, c = 15.37234(9) Å, β = 97.2646(5)°, V = 2427.36(2) Å3, space group P21/n (no. 14), Z = 4, Dc = 1.383 g cm−3, F(000) = 1048.00, T = 223(1) K, μ(Cu-Kα) = 14.374 cm−1, 25682 reflections measured, 4448 independent (Rint = 0.0301). The final refinement converged to R1 = 0.0346 for I > 2.0σ(I), wR2 = 0.0919 for all data.
Yellow solid; M.p. 319.8–321.9 °C; IR (KBr): νmax 1596, 1542, 1517, 1497, 1473, 1454, 1414, 1389, 1352, 1335, 1324, 1238, 1169, 1096, 1069, 1035, 994, 930, 885, 848, 825, 794, 764, 730, 721, 704, 686, 666, 614 cm−1; 1H NMR (500 MHz, CDCl3): δ (ppm) 8.86 (d, J = 7.8 Hz, 1H, Hd), 8.80 (d, J = 8.5 Hz, 1H, Hh), 8.78–7.77 (m, 2H, Hy2), 8.73 (d, J = 7.8 Hz, 1H, Ha), 7.88–7.87 (m, 2H, Hy1), 7.84 (d, J = 7.3 Hz, 1H, Hs), 7.77 (d, J = 7.3 Hz, 1H, Ht), 7.74 (t, J = 7.8 Hz, 1H, Hc), 7.67 (t, J = 7.8 Hz, 1H, Hb), 7.57–7.54 (m, 3H, Hi, Hp1), 7.45–7.40 (m, 3H, Hp2, Hp3), 7.31–7.27 (m, 2H, Hj, Hk); 13C NMR (126 MHz, CDCl3): δ (ppm) 154.3 (Cw), 152.7 (Cv), 150.2 (Cy2), 147.4 (Cq), 144.1 (Cx), 137.9 (Co, Cm), 132.3 (Cu), 132.0 (Cs), 129.64 (Cg), 129.59 (Cp3), 129.56 (Cp2), 128.9 (Cp1), 128.4 (Cf), 128.3 (Cn), 127.8 (Ct), 127.4 (Cc), 127.2 (Ce), 126.4 (Cj), 125.8 (Cb), 125.3 (Ci), 125.0 (Cr), 124.2 (Ch), 123.6 (Cy1), 123.1 (Ca), 122.9 (Cd, Cl), 121.1 (Ck); HRMS-ESI (m/z): [M + H]+ calcd. for C32H20N5S, 506.1434; found, 506.1448. Crystal data for 1 (CCDC 1935319): C32H19N5S, M = 505.60, monoclinic, a = 9.71088(5) Å, b = 16.39216(9) Å, c = 15.37234(9) Å, β = 97.2646(5)°, V = 2427.36(2) Å3, space group P21/n (no. 14), Z = 4, Dc = 1.383 g cm−3, F(000) = 1048.00, T = 223(1) K, μ(Cu-Kα) = 14.374 cm−1, 25682 reflections measured, 4448 independent (Rint = 0.0301). The final refinement converged to R1 = 0.0346 for I > 2.0σ(I), wR2 = 0.0919 for all data.
Preparation of single crystals for cocrystals 1·2a–d
Single crystals of 1·2a were obtained from vapor diffusion of hexane into a chloroform solution (1.4 mL) of a 1:1 molar mixture of 1 (10.2 mg, 0.040 mmol) and 2a (5.1 mg, 0.040 mmol). Other cocrystals 1·2b–d were also obtained by the same procedure.
Crystal data for 1·2a (CCDC 1935320): C39H25N5O2S, M = 627.72, monoclinic, a = 9.93969(8) Å, b = 17.64482(16) Å, c = 17.57204(14) Å, β = 93.1282(8)°, V = 3077.26(4) Å3, space group P21/n (no. 14), Z = 4, Dc = 1.355 g cm−3, F(000) = 1304.00, T = 223(1) K, μ(Cu-Kα) = 12.956 cm−1, 33807 reflections measured, 5609 independent (Rint = 0.0471). The final refinement converged to R1 = 0.0355 for I > 2.0σ(I), wR2 = 0.0942 for all data.
Crystal data for 1·2b (CCDC 1935321): C40H24F3N5O2S, M = 695.72, monoclinic, a = 10.12758(5) Å, b = 16.68174(10) Å, c = 19.48203(10) Å, β = 99.0942(5)°, V = 3250.03(3) Å3, space group P21/n (no. 14), Z = 4, Dc = 1.422 g cm−3, F(000) = 1432.00, T = 223(1) K, μ(Cu-Kα) = 14.217 cm−1, 47704 reflections measured, 5951 independent (Rint = 0.0425). The final refinement converged to R1 = 0.0568 for I > 2.0σ(I), wR2 = 0.1623 for all data.
Crystal data for 1·2c (CCDC 1935322): C40H24N6O2S, M = 652.73, monoclinic, a = 9.72297(7) Å, b = 17.69375(11) Å, c = 18.28982(12) Å, β = 94.3128(6)°, V = 3137.59(4) Å3, space group P21/n (no. 14), Z = 4, Dc = 1.382 g cm−3, F(000) = 1352.00, T = 223(1) K, μ(Cu-Kα) = 13.041 cm−1, 34401 reflections measured, 5741 independent (Rint = 0.0333). The final refinement converged to R1 = 0.0357 for I > 2.0σ(I), wR2 = 0.0942 for all data.
Crystal data for 1·2d (CCDC 1935323): C39H24N6O4S, M = 672.72, monoclinic, a = 9.74862(8) Å, b = 17.91753(15) Å, c = 17.82303(15) Å, β = 92.1261(8)°, V = 3111.03(4) Å3, space group P21/n (no. 14), Z = 4, Dc = 1.436 g cm−3, F(000) = 1392.00, T = 223(1) K, μ(Cu-Kα) = 13.822 cm−1, 36448 reflections measured, 5700 independent (Rint = 0.0452). The final refinement converged to R1 = 0.0389 for I > 2.0σ(I), wR2 = 0.1062 for all data.
Theoretical calculations
The theoretical calculations were performed using the Gaussian 16 program.13 The six lowest singlet–singlet transitions of 1 in 1·2a–d and 1·2a–d were calculated using time-dependent density functional theory (TD-DFT) calculations at the CAM-B3LYP/6-31G(d) level of theory (Table S1†). The molecular structures, obtained from the single-crystal X-ray diffraction analysis, were used as a starting point. Here, the long-range-corrected hybrid functional CAM-B3LYP was used, as CAM-B3LYP often provides better results in TD-DFT calculations than B3LYP, which is conventionally used in DFT calculations.14Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was partly supported by JSPS KAKENHI Grant Number 18H04508 in Grant-in-Aid for Scientific Research on Innovative Areas “Soft Crystals: Area No. 2903”. The authors are grateful to Mr. Shinji Ishihara (Instrumental Analysis Center, Yokohama National University) for carrying out the HRMS-ESI analyses.Notes and references
- For recent reviews, see: (a) M. Kato, H. Ito, M. Hasegawa and K. Ishii, Chem. – Eur. J., 2019, 25, 5105 CrossRef CAS PubMed; (b) Y. Sagara, S. Yamane, M. Mitani, C. Weder and T. Kato, Adv. Mater., 2016, 28, 1073 CrossRef CAS; (c) Z. Ma, Z. Wang, M. Teng, Z. Xu and X. Jia, ChemPhysChem, 2015, 16, 1811 CrossRef CAS.
- (a) P.-F. Feng, M.-Y. Kong, Y.-W. Yang, P.-R. Su, C.-F. Shan, X.-X. Yang, J. Cao, W.-S. Liu, W. Feng and Y. Tang, ACS Appl. Mater. Interfaces, 2019, 11, 1247 CrossRef CAS; (b) X. Wu, J. Guo, W. Jia, J. Zhao, D. Jia and H. Shan, Dyes Pigm., 2019, 162, 855 CrossRef CAS; (c) M. Wang, L. Qian, Y. Guo, H. Wu, M. Liu, W. Gao, G. Li, J. Ding and X. Huang, Dyes Pigm., 2019, 160, 378 CrossRef CAS; (d) S. Li and D. Yan, Adv. Opt. Mater., 2018, 6, 1800445 CrossRef; (e) Q. Benito, C. M. Balogh, H. E. Moll, T. Gacoin, M. Cordier, A. Rakhmatullin, C. Latouche, C. Martineau-Corcos and S. Perruchas, Chem. – Eur. J., 2018, 24, 18868 CrossRef CAS; (f) X.-L. Lv, L.-H. Xie, B. Wang, M. Zhao, Y. Cui and J.-R. Li, J. Mater. Chem. C, 2018, 6, 10628 RSC; (g) C. M. Brown, V. Carta and M. O. Wolf, Chem. Mater., 2018, 30, 5786 CrossRef CAS; (h) Q.-M. Zhou, Z.-R. Deng, S.-S. Yu, L. Wang, H.-B. Duan, Y. Zhou and X.-M. Ren, Eur. J. Inorg. Chem., 2018, 3748 CrossRef CAS; (i) R. R. Cui, Y. C. Lv, Y. S. Zhao, N. Zhao and N. Li, Mater. Chem. Front., 2018, 2, 910 RSC; (j) M. Li, Y. Yuan and Y. Chen, ACS Appl. Mater. Interfaces, 2018, 10, 1237 CrossRef CAS; (k) M. G. S. Londesborough, J. Dolanský, L. Cerdán, K. Lang, T. Jelínek, J. M. Oliva, D. Hnyk, D. Roca-Sanjuán, A. Francés-Monerris, J. Martinčík, M. Nikl and J. D. Kennedy, Adv. Opt. Mater., 2017, 5, 1600694 CrossRef; (l) H. Ohara, T. Ogawa, M. Yoshida, A. Kobayashi and M. Kato, Dalton Trans., 2017, 46, 3755 RSC; (m) A. Pramanik and D. Haldar, RSC Adv., 2017, 7, 389 RSC.
- For seminal examples of MCL dyes, see: (a) Y. Sagara, T. Mutai, I. Yoshikawa and K. Araki, J. Am. Chem. Soc., 2007, 129, 1520 CrossRef CAS PubMed; (b) J. Kunzelman, M. Kinami, B. R. Crenshaw, J. D. Protasiewicz and C. Weder, Adv. Mater., 2008, 20, 119 CrossRef CAS; (c) H. Ito, T. Saito, N. Oshima, N. Kitamura, S. Ishizaka, Y. Hinatsu, M. Wakeshima, M. Kato, K. Tsuge and M. Sawamura, J. Am. Chem. Soc., 2008, 130, 10044 CrossRef CAS PubMed; (d) T. Abe, T. Itakura, N. Ikeda and K. Shinozaki, Dalton Trans., 2009, 711 RSC; (e) Y. Ooyama, Y. Kagawa, H. Fukuoka, G. Ito and Y. Harima, Eur. J. Org. Chem., 2009, 5321 CrossRef CAS; (f) G. Zhang, J. Lu, M. Sabat and C. L. Fraser, J. Am. Chem. Soc., 2010, 132, 2160 CrossRef CAS; (g) S. Perruchas, X. F. L. Goff, S. Maron, I. Maurin, F. Guillen, A. Garcia, T. Gacoin and J.-P. Boilot, J. Am. Chem. Soc., 2010, 132, 10967 CrossRef CAS PubMed; (h) X. Zhang, Z. Chi, H. Li, B. Xu, X. Li, W. Zhou, S. Liu, Y. Zhang and J. Xu, Chem. - Asian J., 2011, 6, 808 CrossRef CAS PubMed; (i) X. Zhang, Z. Chi, J. Zhang, H. Li, B. Xu, X. Li, S. Liu, Y. Zhang and J. Xu, J. Phys. Chem. B, 2011, 115, 7606 CrossRef CAS PubMed; (j) S.-J. Yoon and S. Y. Park, J. Mater. Chem., 2011, 21, 8338 RSC; (k) Y. Dong, B. Xu, J. Zhang, X. Tan, L. Wang, J. Chen, H. Lv, S. Wen, B. Li, L. Ye, B. Zou and W. Tian, Angew. Chem., Int. Ed., 2012, 51, 10782 CrossRef CAS PubMed; (l) K. Nagura, S. Saito, H. Yusa, H. Yamawaki, H. Fujihisa, H. Sato, Y. Shimoikeda and S. Yamaguchi, J. Am. Chem. Soc., 2013, 135, 10322 CrossRef CAS PubMed; (m) M. Krikorian, S. Liu and T. M. Swager, J. Am. Chem. Soc., 2014, 136, 2952 CrossRef CAS.
- Little is known about MCL dyes that exhibit multi-color switching. For examples, see: (a) S. Ito, G. Katada, T. Taguchi, I. Kawamura, T. Ubukata and M. Asami, CrystEngComm, 2019, 21, 53 RSC; (b) X. Wu, J. Guo, Y. Cao, J. Zhao, W. Jia, Y. Chen and D. Jia, Chem. Sci., 2018, 9, 5270 RSC; (c) W. Yang, C. Liu, S. Lu, J. Du, Q. Gao, R. Zhang, Y. Liu and C. Yang, J. Mater. Chem. C, 2018, 6, 290 RSC; (d) M. Okazaki, Y. Takeda, P. Data, P. Pander, H. Higginbotham, A. P. Monkman and S. Minakata, Chem. Sci., 2017, 8, 2677 RSC; (e) S. Ito, T. Yamada and M. Asami, ChemPlusChem, 2016, 81, 1272 CrossRef; (f) Z. Ma, Z. Wang, X. Meng, Z. Ma, Z. Xu, Y. Ma and X. Jia, Angew. Chem., Int. Ed., 2016, 55, 519 CrossRef CAS PubMed; (g) Y. Sagara and T. Kato, Angew. Chem., Int. Ed., 2011, 50, 9128 CrossRef CAS PubMed.
- For recently reported examples of multi-stimuli-responsive MCL dyes, see: (a) X. Liu, M. Li, M. Liu, Q. Yang and Y. Chen, Chem. – Eur. J., 2018, 24, 13197 CrossRef CAS PubMed; (b) Q. Hou, L. Liu, S. K. Mellerup, N. Wang, T. Peng, P. Chen and S. Wang, Org. Lett., 2018, 20, 6467 CrossRef CAS PubMed; (c) P.-Z. Chen, H. Zhang, L.-Y. Niu, Y. Zhang, Y.-Z. Chen, H.-B. Fu and Q.-Z. Yang, Adv. Funct. Mater., 2017, 27, 1700332 CrossRef; (d) Q. Kong, W. Zhuang, G. Li, Y. Xu, Q. Jiang and Y. Wang, New J. Chem., 2017, 41, 13784 RSC; (e) L. Liu, X. Wang, N. Wang, T. Peng and S. Wang, Angew. Chem., Int. Ed., 2017, 56, 9160 CrossRef CAS PubMed; (f) B. Xu, H. Wu, J. Chen, Z. Yang, Z. Yang, Y.-C. Wu, Y. Zhang, C. Jin, P.-Y. Lu, Z. Chi, S. Liu, J. Xu and M. Aldred, Chem. Sci., 2017, 8, 1909 RSC; (g) T. Jadhav, B. Dhokale, Y. Patil, S. M. Mobin and R. Misra, J. Phys. Chem. C, 2016, 120, 24030 CrossRef CAS; (h) C. Jobbágy, P. Baranyai, G. Marsi, B. Rácz, L. Li, P. Naumov and A. Deák, J. Mater. Chem. C, 2016, 4, 10253 RSC; (i) K. Yang, S.-L. Li, F.-Q. Zhang and X.-M. Zhang, Inorg. Chem., 2016, 55, 7323 CrossRef CAS PubMed; (j) R. Hagihara, N. Harada, S. Karasawa and N. Koga, CrystEngComm, 2015, 17, 8825 RSC.
- For recently reported examples of solid-state fluorophores with independent HCL/MCL systems, see: (a) F. Wang, D. Song, D. A. Dickie and C. L. Fraser, Chem. - Asian J., 2019, 14, 1849 CrossRef CAS PubMed; (b) B. Wang and C. Wei, RSC Adv., 2018, 8, 22806 RSC; (c) A. Ekbote, S. M. Mobin and R. Misra, J. Mater. Chem. C, 2018, 6, 10888 RSC; (d) J. Xiong, K. Wang, Z. Yao, B. Zou, J. Xu and X.-H. Bu, ACS Appl. Mater. Interfaces, 2018, 10, 5819 CrossRef CAS PubMed; (e) W. Fang, W. Zhao, P. Pei, R. Liu, Y. Zhang, L. Kong and J. Yang, J. Mater. Chem. C, 2018, 6, 9269 RSC; (f) S. Chen, W. Liu, Z. Ge, W. Zhang, K.-P. Wang and Z.-Q. Hu, CrystEngComm, 2018, 20, 5432 RSC; (g) B. Roy, M. C. Reddy and P. Hazra, Chem. Sci., 2018, 9, 3592 RSC; (h) Z. Wang, X. Cheng, A. Qin, H. Zhang, J. Z. Sun and B. Z. Tang, J. Phys. Chem. B, 2018, 122, 2165 CrossRef CAS PubMed; (i) F. Wang, C. A. DeRosa, M. L. Daly, D. Song, M. Sabat and C. L. Fraser, Mater. Chem. Front., 2017, 1, 1866 RSC; (j) W. Fang, Y. Zhang, G. Zhang, L. Kong, L. Yang and J. Yang, CrystEngComm, 2017, 19, 1294 RSC; (k) X.-L. Lu and M. Xia, J. Mater. Chem. C, 2016, 4, 9350 RSC.
- (a) M. Kondo, T. Yamoto, S. Miura, M. Hashimoto, C. Kitamura and N. Kawatsuki, Chem. - Asian J., 2019, 14, 471 CrossRef CAS; (b) P. Xue, P. Chen, J. Jia, Q. Xu, J. Sun, B. Yao, Z. Zhang and R. Lu, Chem. Commun., 2014, 50, 2569 RSC.
- (a) S. Nagai, M. Yamashita, T. Tachikawa, T. Ubukata, M. Asami and S. Ito, J. Mater. Chem. C, 2019, 7, 4988 RSC; (b) S. Ito, T. Taguchi, T. Yamada, T. Ubukata, Y. Yamaguchi and M. Asami, RSC Adv., 2017, 7, 16953 RSC; (c) S. Ito, T. Yamada, T. Taguchi, Y. Yamaguchi and M. Asami, Chem. - Asian J., 2016, 11, 1963 CrossRef CAS PubMed.
- For examples of MCL-active benzothiadiazole derivatives, see: (a) T. Ishi-i, H. Tanaka, R. Youfu, N. Aizawa, T. Yasuda, S. Kato and T. Matsumoto, New J. Chem., 2019, 43, 4998 RSC; (b) G. He, L. Du, Y. Gong, Y. Liu, C. Yu, C. Wei and W. Z. Yuan, ACS Omega, 2019, 4, 344 CrossRef CAS; (c) C.-Y. Yu, C.-C. Hsu and H.-C. Weng, RSC Adv., 2018, 8, 12619 RSC; (d) A. Ekbote, S. M. Mobin and R. Misra, J. Mater. Chem. C, 2018, 6, 10888 RSC; (e) J. Chen, D. Li, W. Chi, G. Liu, S. H. Liu, X. Liu, C. Zhang and J. Yin, Chem. – Eur. J., 2018, 24, 3671 CrossRef CAS PubMed; (f) L. Pan, Y. Cai, H. Wu, F. Zhou, A. Qin, Z. Wang and B. Z. Tang, Mater. Chem. Front., 2018, 2, 1310 RSC; (g) K. C. Naeem, K. Neenu and V. C. Nair, ACS Omega, 2017, 2, 9118 CrossRef CAS.
- For examples of optoelectronic properties of phenanthroimidazole derivatives, see: (a) N. Zhao, Y. Li, Y. Jia and P. Li, J. Phys. Chem. C, 2018, 122, 26576 CrossRef CAS; (b) Y. Li, W. Wang, Z. Zhuang, Z. Wang, G. Lin, P. Shen, S. Chen, Z. Zhao and B. Z. Tang, J. Mater. Chem. C, 2018, 6, 5900 RSC; (c) V. Thanikachalam, P. Jeeva and J. Jayabharathi, RSC Adv., 2017, 7, 13604 RSC; (d) W.-C. Chen, Y. Yuan, S.-F. Ni, Q.-X. Tong, F.-L. Wong and C.-S. Lee, Chem. Sci., 2017, 8, 3599 RSC; (e) K. Skonieczny, J. Yoo, J. M. Larsen, E. M. Espinoza, M. Barbasiewicz, V. I. Vullev, C.-H. Lee and D. T. Gryko, Chem. – Eur. J., 2016, 22, 7485 CrossRef CAS PubMed; (f) K. Wang, S. Wang, J. Wei, S. Chen, D. Liu, Y. Liu and Y. Wang, J. Mater. Chem. C, 2014, 2, 6817 RSC; (g) R. Francke and R. D. Little, J. Am. Chem. Soc., 2014, 136, 427 CrossRef CAS PubMed.
- It should be noted that the green-emissive crystals of 1 changed to non-emissive states upon exposure to vapors of hydrochloric acid, acetic acid, or trifluoroacetic acid.
- (a) A. P. Harding, D. C. Wedge and P. L. A. Popelier, J. Chem. Inf. Model., 2009, 49, 1914 CrossRef CAS; (b) G.-l. Li and G. Zhao, J. Org. Chem., 2005, 70, 4272 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision A.03, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- D. Jacquemin, E. A. Perpète, G. E. Scuseria, I. Ciofini and C. Adamo, J. Chem. Theory Comput., 2008, 4, 123 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Spectral data, X-ray diffraction analyses, theoretical calculations, DSC analyses, and PXRD data. CCDC reference numbers: 1935319 (1), 1935320 (1·2a), 1935321 (1·2b), 1935322 (1·2c), and 1935323 (1·2d). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9ce01037h |
| This journal is © The Royal Society of Chemistry 2019 |