
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Tuning diffusion paths in shaped ceria nanocrystals†
Vincenzo
Esposito
 *a,
Ahsanul
Kabir
a,
Massimo
Rosa
a,
Ngo
Van Nong
a,
Thenner S.
Rodrigues
b,
Lays N.
Rodrigues
b,
Marina F. S.
Machado
b,
Letícia P. R.
Moraes
b,
Debora
Marani
c and
Fabio Coral
Fonseca
b
*a,
Ahsanul
Kabir
a,
Massimo
Rosa
a,
Ngo
Van Nong
a,
Thenner S.
Rodrigues
b,
Lays N.
Rodrigues
b,
Marina F. S.
Machado
b,
Letícia P. R.
Moraes
b,
Debora
Marani
c and
Fabio Coral
Fonseca
b
aTechnical University of Denmark, Department of Energy Conversion and Storage, Frederiksborgvej 399, DK-4000 Roskilde, Denmark. E-mail: vies@dtu.dk
bInstituto de Pesquisas Energéticas e Nucleares – IPEN-CNEN/SP, São Paulo, SP 05508-000, Brazil
cCentro de Engenharia, Modelagem e Ciências Sociais Aplicadas, Universidade Federal do ABC, Santo André, (SP) 09210-580, Brazil
First published on 17th June 2019
Abstract
Mass diffusion controls material structuring from the atomic to the macro-scale defining properties and functionalities. We show here that surface energy in Ce0.9Gd0.1O3-d shaped nanocrystals, i.e. nanocubes, nanorods and spherical nanoparticles, can control solid state diffusion mechanisms over a long range, leading to extreme microstructural diversity.
Solid state mass diffusion (SSD) is crucial in materials technologies. As dictated by Fick's laws, SSD controls the migration of atoms in space and time, occurring exclusively at the solid state via interface and bulk crystalline paths.1 SSD regulates material formation into their final shapes, from the atomic to the macro-scale. Diffusion thus regulates the final microstructure, e.g., by regulating grain size and porosity in sintering. SSD also controls mechanical performances at high temperatures, as in creep, in solid to solid joining, and chemical processes such as solid–solid chemical contamination, powder processing and doping.1
The SSD process in oxides is thermally activated and influenced by the composition, defects, and oxygen gas activity and by the morphology of the starting materials.2–5 Morphology is particularly relevant to control the final microstructures, especially when the starting materials are on the nanometric scale. Nanomaterials can rapidly grow with increasing temperature and time and can even lead to crystallization of three-dimensional superlattices mimicking the morphology on the starting nanometric units.4 Size also matters in SSD and microstructures rapidly change from the nano- to the micro-scale through fast diffusive mechanisms.1–6 Nanomaterials can indeed densify fast at lower temperatures because of high surface energy contribution in the mass diffusion process. This occurs mainly for highly packed nanoparticles at the green stage, where fast diffusion activates simultaneous necking among the particles. However, size effects can also impact SSD in alternative ways. SSD mechanisms are rather complex: slow and fast mechanisms can compete in nanoparticles with significantly different results.3,7–9 The morphological features of nanomaterials can rapidly evolve via mass flow, dissipating the high surface energy contribution at the nanoparticles, thus creating preferential paths of diffusion, controlled by slow mechanisms, and thus leaving densification incomplete.6 As fast necking between nanoparticles can be easily activated at low temperature, it can also favour agglomeration, inhibiting densification at high temperatures.1,6,10 Particularly for the so-called bulk diffusive mechanism/s, activated above 0.7Tfusion (e.g., for the last stage of the sintering), a long range crystalline order is needed to transport mass from the core of a crystal to its neighbours.1,6 The disorder introduced by the large grain boundary generally inhibits such mechanisms.11 Therefore, while low activation energy is required to activate the mass diffusion at the interfaces of nanomaterials (surface and grain boundaries), intense mass transport within and between large crystals through the interface barrier requires high thermal energy.6,11 In either cases, the interface plays a key role as the diffusion takes place at the contact between particles.
In this study, we discriminate the surface SSD mechanisms by comparing the thermal evolution of nanocrystals with defined crystalline morphologies. We especially investigate 10% molar gadolinium-doped ceria (GDC) for its relevance in several chemical, environmental and energy technologies, and for the knowledge gathered on its diffusion effects.11–16 For the morphologies, we synthesized GDC as nanocubes (NCs) and nanorods (NRs) by hydrothermal synthesis, and random spherical nanoparticles (RDs) were obtained by co-precipitation. The chemical methods are described in detail in ref. 17 and 6, respectively.
Fig. 1 shows the overall structural (X-ray diffraction, XRD) patterns (left hand side) and microstructural features observed by both scanning electron microscopy (SEM) and transmission electron microscopy (TEM) of the starting materials after calcination at 450 °C for 2 h. The XRD data show that the starting materials are crystalline, with a typical fluorite diffraction pattern. The XRD analysis also clearly indicates rather broad diffraction peaks for both the RD and the NR samples, while the NC pattern exhibits narrow peaks. These peak widths correspond to average crystallite sizes, calculated by the Scherrer equation, of ca. 9, 47, and 7 nm for RDs, NCs and NRs, respectively. As shown in the SEM and TEM images in Fig. 1, such differences at the structural level are also reflected at the microstructural and crystalline levels. Particularly for the NCs, the higher intensity of the diffraction peaks is attributed to the sharp cubic morphology of the fluorite symmetry that ranges in tens of nanometers. The NRs exhibit a linear extension of ca. 100 nm and a section in the order of a few nanometers (ca. 10 nm) with a rather narrow distribution of sizes. The RD nanoparticles have an average size of a few tens of nanometers and the TEM inset indicates a dominant spheroidal shape with no preferential crystallographic orientation.
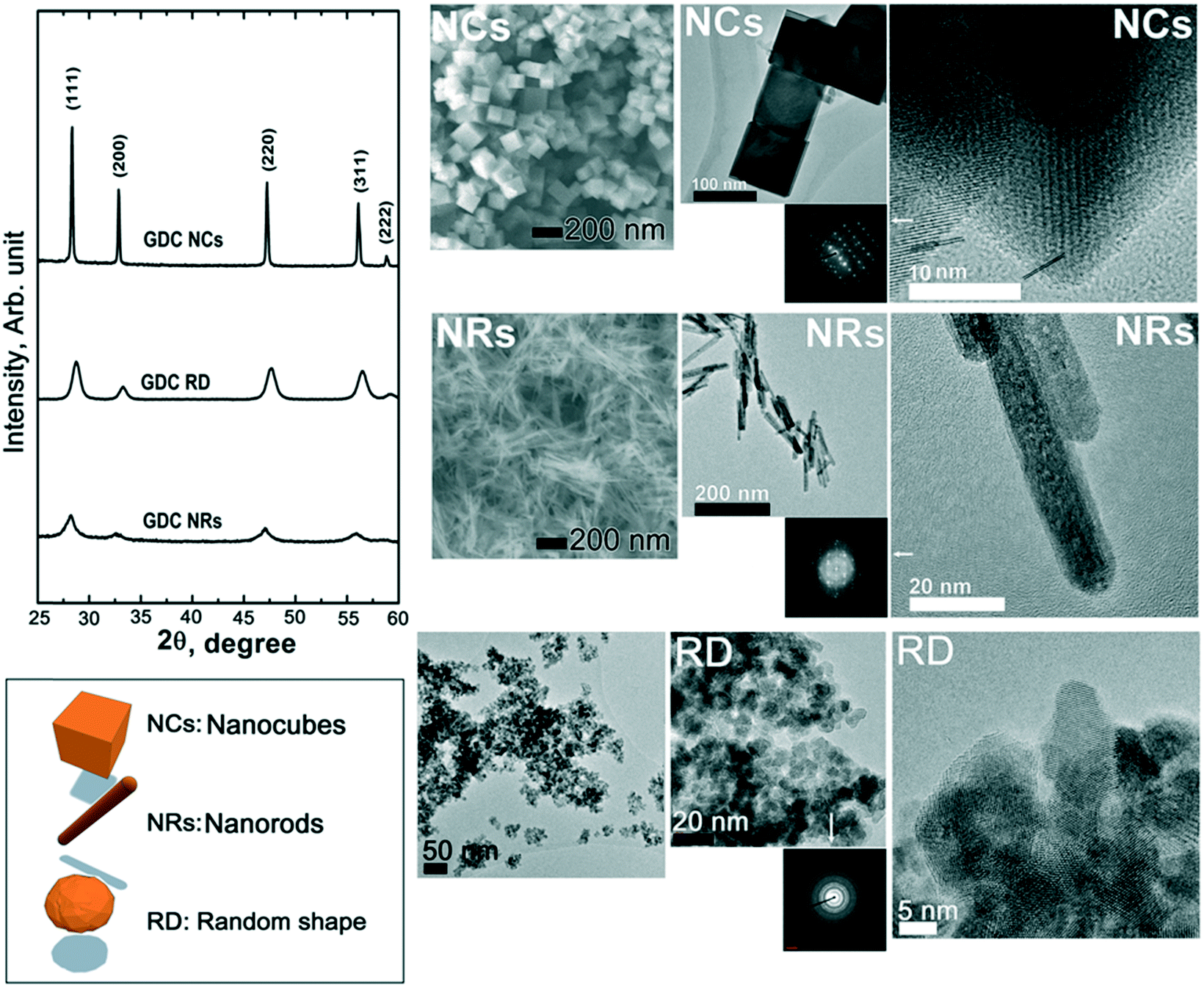 | ||
| Fig. 1 Structural X-ray diffraction patterns (left hand side) and microstructural features observed by both scanning electron microscopy (SEM) and transmission electron microscopy (TEM) of the starting materials after calcination at 450 °C for 2 h. | ||
The thermodynamic behaviour of nanocrystals is mainly associated with the energetic term (γ × A), where γ and A are the surface free energy and surface area, respectively.18 The A values can be evaluated by simply applying the formulas of the corresponding geometrical shapes, whereas the γ values can be obtained in previously published data. For instance, previous reports indicated an average value of 0.3 J m−2 for random ceria nanoparticles.2 For the NCs, the characteristic surface energy of the (100), (110) and (111) faces is expected to be as high as ∼1.6 J m−2 (as for the relaxed crystals in ref. 19), equivalent for all the six faces of the cubes. The NR sample shows two main orientations along the 1-D extension, where the crystals are expected to minimize the surface energy to 0.3–0.7 J m−2.19 Considering the different shapes observed from the TEM images and the corresponding nanocrystal dimensions, the energetic content at the surfaces of the individual nanocrystals is expected to be: RDs = 0.3 × Ssphere = 76.3 × 10−18 J, NCs = 1.6 × Scube = 21.2 × 10−15 J, and NRs = 0.5 × Srods = 25.3 × 10−16 J. Among the three differently shaped nanocrystals, we can thus estimate the maximum surface energy content of the NCs. Despite the calculations, the NC sample reflects the fluorite symmetry, possessing surfaces with the same chemical energy and sharp edges with a high curvature. The edges are thus expected to be highly reactive for the NCs and NRs, while the spherical particles will become reactive mainly when in contact with neighbouring particles, as per the Coble model.2 Such anisotropy in the NCs can in principle induce the formation of preferential surface diffusion paths, where the crystals are packed by edge–edge, face–face or surface–edge contacts.
Fig. 2 shows the contact dilatometry (Setaram Instrumentation) curves and SEM images of the sintered samples at 1400 °C. The NC, NR and RD samples are pressed under the same conditions (uniaxial pressure of 150 MPa) with relative green densities (ρ0) in the 40–60% range depending on the particle shape (Fig. 2(a)). A high green density (ρ0 > 50%) was achieved for both NCs and RDs, while a slightly lower value (<45%) was found for NRs. The ρ0 values were measured from several samples confirming that the initial powder shape influences particle packing. The observed differences are attributed to the shape-driven packing process, which, along with possible agglomeration effects for the RD sample, results in a different particle-to-particle coordination in the green ceramic body depending on the shape of the crystals.3,4,6 For the diffusion leading to the densification process (Fig. 2(b)), the green density is a crucial parameter that controls the densification rate as well as the residual porosity under the final equilibrium conditions.3,6 According to Chen et al., loosely packed starting particles are generally expected to remain porous at high temperature.2 The SSD is monitored by free sintering (pressure-free) with a constant heating rate of 10 °C min−1 in air (synthetic air flow at 50 ml min−1). The experimental data are presented here as the densification (Fig. 2a) and densification rate (Fig. 2b) as a function of the temperature. The densification rate is calculated as reported in previous studies to represent diffusion activity as a function of the thermal energy (see ref. 11–14 and 20). The plots display several unexpected features. Generally, the fastest densification is expected for the highest ρ0, i.e. the NCs (ρ0-NC = 0.55). However, the NRs, with the lowest green density (ρ0-NR = 0.4), show the fastest densification starting at low temperatures between 300 and 500 °C. Fig. 2 also reveals that NCs exhibit no densification but rather a slight expansion during the thermal treatment. By analysing the dρ dT−1 curve, dilatometry indicates that the diffusive activity leading to densification is practically null for the NCs. Conversely, diffusion is well-pronounced for the NRs. Interestingly the rods show an accelerated diffusive activity with two local maxima taking place at 800 and 1050 °C, where the intensity of the mass diffusion at 1050 °C is higher than that at 800 °C. This second high-temperature maximum is also unexpected and it indicates a two-stage diffusive process, where the first stage controls the second one. Such a diffusive behaviour is typical of multiphase systems such as composite or porous materials.9 Conversely for the RD powders, a typical densification process consists of a progressive, single-step densification, where the most intense diffusive activity activates at the first stage (i.e. the particle necking and rapid densification of the nanoparticles). The intermediate stage of the sintering, corresponding to residual porosity annihilation and grain growth, has generally minor densification, i.e. in the RD sample for temperatures above 900 °C. The SEM analysis of the samples after a heat treatment of 0.1 hours and after a prolonged sintering of 10 hours at 1400 °C is reported in Fig. 2(right panel). The final microstructures indicate a nearly full densification of both the NR and RD samples, with the typical polycrystalline morphology and flat grain boundary. A careful analysis of grain boundaries in the sintered NRs indicates a fast and complete diffusive process that resulted in a rather homogeneous microstructure with a narrow grain size distribution and no traces of stable porosity, as expected from the two-stage activity in Fig. 2b. For the RD sample, some residual porosity could be encountered as a result of typical agglomeration issues often found in the fast sintering of nanoparticles (not shown in Fig. 2).6 As for the most interesting result, it is seen in the NC solid state diffusion process, in which open porosity is structured in the body of the samples after both heat treatments at 0.1 and 10 hours. The rounded shapes of grains after sintering for 10 hours in the NC sample indicate an intense diffusive process that promotes the change from the cubic shape to the spherical shape. Remarkably, the grains have well-defined and limited necking and this feature leads to extensive rounded porosity in the consolidated ceramic body (digital evaluation of grain and pore sizes was carried out using Image J as in ref. 11). Such a result suggests that the porosity is stable in the NC sample, despite the pores being smaller than the grains, e.g. (∼1 μm pores vs. ∼2 μm grains after 10 hours). The evolution of the crystallite size of both NRs and NCs was measured by XRD analyses of the as-synthesized materials and after heat treatments between 850 °C and 1400 °C (see Fig. S1 in the ESI†). The results showed that SSD in NC crystals is substantially inert, whereas NRs present a rapid evolution of the crystallinity. The diffusion coefficients calculated at 1400 °C, D1400, through the grain boundary as in ref. 2, are DNCs = 3 × 10−14 cm2 s−1, DNRs = 3 × 10−13 cm2 s−1 and DRDs = 5 × 10−13 cm2 s−1. Such estimated values are consistent with previous calculations on GDC at the late stage of sintering and indicate that NCs are at least one order of magnitude less diffusive than RDs and NRs.6,12 Moreover, from the microstructural observations in Fig. 2, the diffusive mechanisms leading to the transition from the specific nanostructured shape to the final sintered grains appear to be controlled by the cubic shape only. On the other hand, NRs lose rapidly the rod-like shape to evolve into spherical particles. SEM characterization of the samples treated at intermediate stages of sintering (800 °C and 1100 °C for 0.1 h) is shown in Fig. S2† and confirms such features. To have a closer look on the microstructural evolution of the nanopowders, a spark plasma sintering (SPS) treatment was carried out at 70 MPa up to 980 °C with 5 min dwelling under a constant dc current of 1 kA, in a vacuum (Dr. Sinter Lab 515S, Fuji-SPS, Japan) on both the NC and NR samples. By such a rapid sintering process, it is possible to have an insight into the preferential and most energetic mass diffusion mechanisms activated under the effect of both the applied electric field and pressure. This is especially relevant for the NC samples that exhibit a slow mass diffusion and stable porosity. Fig. 3 shows the details of the densification profile during SPS (top) and the resulting microstructures by SEM after the treatment of the NC and NR samples (bottom). The SPS profile indicates similar features of the conventional sintering as observed in the dilatometry curves reported in Fig. 2. The SPS profile in Fig. 3 confirms an inhibited mass diffusion transport in the NCs, despite the intense conditions of SPS. Surprisingly, the final relative density of the NC sample was around 50% with half of the sample remaining porous. For the NRs, the SPS process showed fast densification at the early stages. The SEM images in Fig. 3(bottom) clearly show a rapid evolution of the NRs into the spherical grains with very few residual rods in the sample. This result is consistent with the free sintering process (Fig. 2), in which no traces of rods resulted for the sample densified at 1400 °C. On the other hand, Fig. 3 indicates that the NC sample retained its cubic shape after SPS, especially for the few large particles still present in the material (inset Fig. 3).
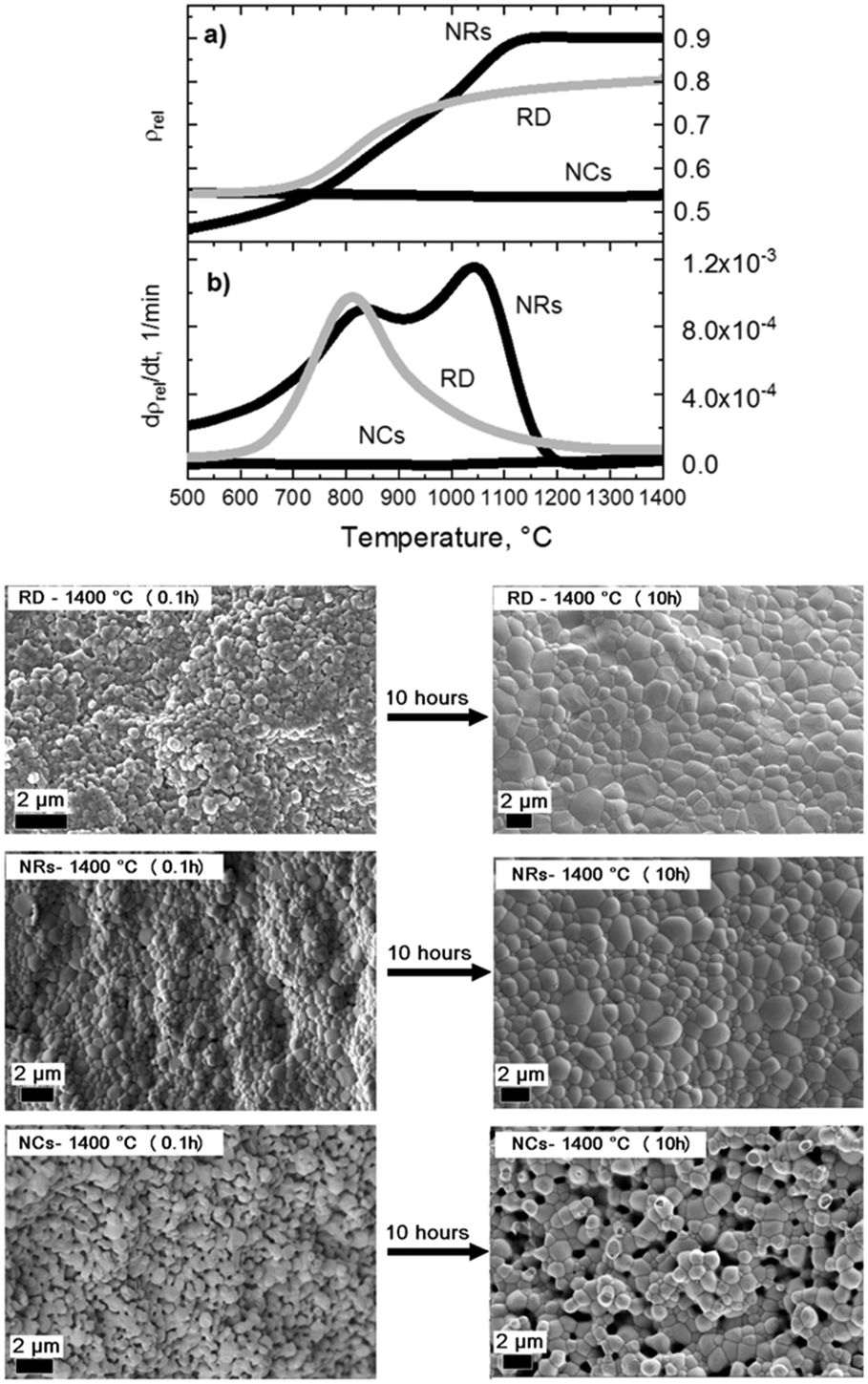 | ||
| Fig. 2 (Top) Densification profile as relative volumetric shrinkage (a) and densification rate (b); (Bottom) SEM images of the sintered samples at 1400 °C for 0.1 (bottom-left) and 10 hours (bottom-right). | ||
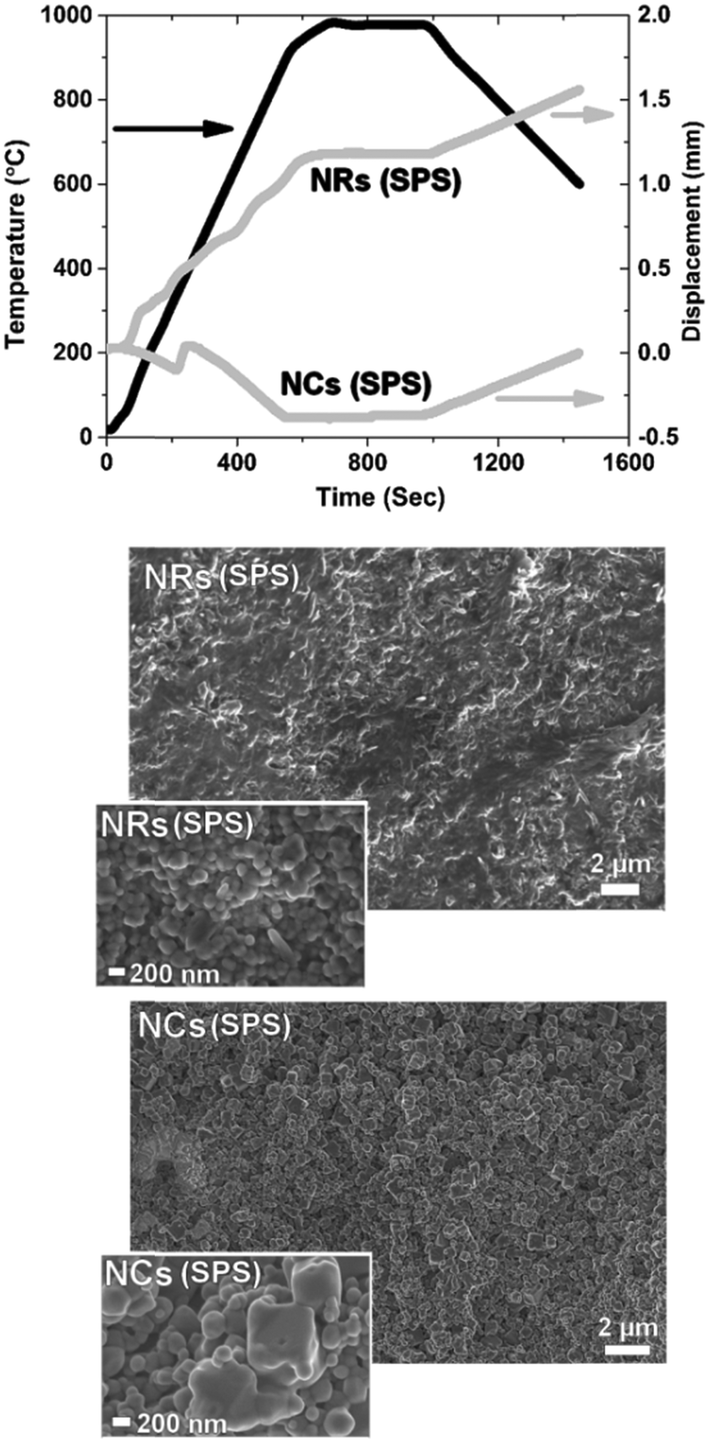 | ||
| Fig. 3 Densification profile during SPS (top) and the resulting microstructures by SEM after the treatment of the NC and NR samples (bottom). | ||
Conclusions
GDC nanoparticles with different shapes have rather dissimilar diffusive behaviours. Such effects are the result of the critical role of the surface mechanisms over the bulk diffusion mechanisms. The preferential surface diffusive paths can control the overall diffusion, even overcoming the packing factors at the green state and the overall energy content in materials, e.g. as expected from the grain size. As an extreme case, we disclose that solid state diffusion mechanisms in nanocubes are geometrically confined, inhibiting porosity annihilation, grain growth and even morphological changes from the cubic shape. The surface energy in NCs leads to a “locked” diffusion, even under the severe conditions of the SPS treatment, with the possibility to form a highly porous continuum with stable porosity, even at high temperatures. Such a result is relevant for those applications in which porous ceria is desired under harsh operative conditions, e.g. in high temperature catalysis, solid oxide electrochemical cells and thermochemical water splitting. Conversely, the high surface energy in NRs leads to a rapid rod to spheroidal transformation and to a rapid densification with maximum activity at ca. 1100 °C. Full density and equilibrium microstructures resulted in NRs, despite the low green density of ca. 45%, overcoming the general effects associated with the particle coordination and densification paths. These two cases strongly suggest that topological features in nanometric ceria can influence the microstructural assembly even in the case of randomly packed particles.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project has partially received funding from the Fuel Cells and Hydrogen 2 Joint Undertaking under Grant agreement No 700266. This Joint Undertaking receives support from the European Union's Horizon 2020 research and innovation program and Hydrogen Europe and N.ERGHY. This work was supported by FAPESP [grant 2014/09087-4, scholarships (2017/04929-5 and 2019/04499-6), RCGI Shell (ANP)/FAPESP (grant 2014/50279-4), and CINE-Shell (ANP)/FAPESP (grant 2017/11937-4)]. Authors V. E., L. P. R. M., D. M., and F. C. F. acknowledge the CNPQ for fellowships (401218/2014-7, 454227/2017-5, and 306258/2016-1). Thanks are also due to CNEN for financial support. The authors also gratefully acknowledge the Danish Council for Independent Research | Technology and Production Sciences for the DFF-Research Project 2 (grant no. 48293) which financially supported the activity of Vincenzo Esposito and Ahsanul Kabir.References
- A. Fick, Ann. Phys., 1855, 170, 59 CrossRef.
- P.-L. Chen and I.-W. Chen, J. Am. Ceram. Soc., 1996, 79, 3129 CrossRef CAS.
- P.-L. Chen and I.-W. Chen, J. Am. Ceram. Soc., 1997, 80, 637 CrossRef CAS.
- L. Wu, J. J. Willis, I. S. MacKay, B. T. Diroll, J. Quin, M. Cargnello and C. J. Tassone, Nature, 2017, 548, 197 CrossRef CAS PubMed.
- F. Teocoli and V. Esposito, Scr. Mater., 2014, 75, 82 CrossRef CAS.
- V. Esposito and E. Traversa, J. Am. Ceram. Soc., 2008, 91, 1037 CrossRef CAS.
- R. Guo, W. Cao, X. Mao and J. Li, J. Am. Ceram. Soc., 2016, 99, 3556 CrossRef CAS.
- F. Teocoli, D. W. Ni, K. Brodersen, S. P. V. Foghmoes, S. Ramousse and V. Esposito, J. Mater. Sci., 2014, 49, 5324 CrossRef CAS.
- D. W. Ni, C. G. Schmidt, F. Teocoli, A. Kaiser, K. B. Andersen, S. Ramousse and V. Esposito, J. Eur. Ceram. Soc., 2013, 33, 2529 CrossRef CAS.
- J. A. Glasscock, V. Esposito, S. P. V. Foghmoes, T. Stegk, D. Matuschek, M. W. H. Ley and S. Ramousse, J. Eur. Ceram. Soc., 2013, 33, 1289 CrossRef CAS.
- V. Esposito, D. W. Ni, Z. He, W. Zhang, A. S. Prasad, J. A. Glasscock, C. Chatzichristodoulou, S. Ramousse and A. Kaiser, Acta Mater., 2013, 61, 6290 CrossRef CAS.
- D. W. Ni, D. Z. de Florio, D. Marani, A. Kaiser, V. B. Tinti and V. Esposito, J. Mater. Chem. A, 2015, 3, 18835 RSC.
- F. Teocoli, D. W. Ni and V. Esposito, Scr. Mater., 2015, 94, 13 CrossRef CAS.
- D. W. Ni, J. A. Glasscock, A. Pons, W. Zhang, A. Prasad, S. Sanna, N. Pryds and V. Esposito, J. Electrochem. Soc., 2014, 161, F3072 CrossRef CAS.
- V. Esposito, D. W. Ni, S. Sanna, F. Gualandris and N. Pryds, RSC Adv., 2017, 7, 13784 RSC.
- V. Esposito, D. W. Ni, D. Marani, F. Teocoli, K. T. Sune Thydén, D. Z. De Florio and F. C. Fonseca, J. Mater. Chem. A, 2016, 4, 16871 RSC.
- T. S. Rodrigues, A. B. L. de Moura, F. A. e Silva, E. G. Candido, A. G. M. da Silva, D. C. de Oliveira, J. Quiroz, P. H. C. Camargo, V. S. Bergamaschi, J. C. Ferreira, M. Linardi and F. C. Fonseca, Fuel, 2019, 237, 1244 CrossRef CAS.
- H. M. Lu and Q. Jiang, J. Phys. Chem. B, 2004, 108, 5617 CrossRef CAS.
- G. I. Nkou Bouala, N. Clavier, S. Martin, J. Léchelle, J. Favrichon, H. P. Brau, N. Dacheux and R. Podor, J. Phys. Chem. C, 2016, 120, 386 CrossRef CAS.
- D. W. Ni, C. G. Schmidt, F. Teocoli, A. Kaiser, K. B. Andersen, S. Ramousse and V. Esposito, J. Eur. Ceram. Soc., 2013, 33, 2529 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ce00414a |
| This journal is © The Royal Society of Chemistry 2019 |