
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Living in the salt-cocrystal continuum: indecisive organic complexes with thermochromic behaviour†
Charlotte L.
Jones
a,
Jonathan M.
Skelton
ab,
Stephen C.
Parker
 ab,
Paul R.
Raithby
*a,
Aron
Walsh
c,
Chick C.
Wilson
*a and
Lynne H.
Thomas
*a
ab,
Paul R.
Raithby
*a,
Aron
Walsh
c,
Chick C.
Wilson
*a and
Lynne H.
Thomas
*a
aDepartment of Chemistry, University of Bath, Claverton Down, Bath BA2 7AY, UK. E-mail: p.r.raithby@bath.ac.uk; c.c.wilson@bath.ac.uk; l.h.thomas@gmail.com
bSchool of Chemistry, University of Manchester, Oxford Road, Manchester M13 9PL, UK
cDepartment of Materials, Imperial College, London, Exhibition Road, London SW7 2AZ, UK
First published on 7th February 2019
Abstract
A family of multicomponent haloaniline/3,5-dinitrobenzoic acid molecular crystals with striking red-to-colourless temperature-induced thermochromism is identified and characterised. Four thermochromic pairs of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 neutral cocrystals and ionic salts are identified which, unusually, grow concomitantly under the same conditions. The coloured cocrystals are found to be metastable, kinetically trapped during crystallisation, and convert via proton transfer to the more stable salt forms on heating. The colour of the neutral form and the temperature of the transition can be tuned through the halogen and by chemical substitution on the aniline component. From structural characterisation and first-principles modelling, we elucidate the origin of the metastability of the cocrystals and link structural changes through the phase transition to the striking visible colour change. By deliberately exploiting the uncertainty of the salt-cocrystal continuum, where the small pKa difference between components enables significant solid-state structural rearrangements induced by proton transfer, this work highlights a novel design paradigm for engineering new organic thermochromics with tailored physical properties.
:1 neutral cocrystals and ionic salts are identified which, unusually, grow concomitantly under the same conditions. The coloured cocrystals are found to be metastable, kinetically trapped during crystallisation, and convert via proton transfer to the more stable salt forms on heating. The colour of the neutral form and the temperature of the transition can be tuned through the halogen and by chemical substitution on the aniline component. From structural characterisation and first-principles modelling, we elucidate the origin of the metastability of the cocrystals and link structural changes through the phase transition to the striking visible colour change. By deliberately exploiting the uncertainty of the salt-cocrystal continuum, where the small pKa difference between components enables significant solid-state structural rearrangements induced by proton transfer, this work highlights a novel design paradigm for engineering new organic thermochromics with tailored physical properties.
Introduction
Dynamic solid-state materials have found diverse applications including as sensors and switches. Colour and colour changes in response to environmental stimuli are attractive target properties, as the property change can be easily detected and correlated to structural changes. Colour switching may be accessed through a range of external stimuli including light,1 temperature2–4 and pressure.5 Most efforts to design new functional materials in this domain focus on inorganic metal-containing compounds, but organic solids are arguably a more attractive option, being lightweight, processable and cost effective, and thus desirable targets for many applications.3,4Given their crystalline nature, controlling molecular assembly via crystal-engineering techniques is the obvious route to modifying the physical properties of organic solids.6 While this technique has been widely used in areas such as pharmaceuticals,7–9 studies focussing on manipulating optical properties are more limited, although reported examples include non-linear optical10,11 and luminescent12–15 materials. One route to modified properties is the formation of multi-component complexes, where the introduction of a second molecule into the crystal lattice can facilitate the engineering of solid-state forms with significantly different properties.16–18
In this work, we sought to develop switchable materials by creating and switching between neutral cocrystals and ionic salts. In cocrystals, both components are formally neutral and typically interact through designable intermolecular interactions such as hydrogen or halogen bonding. Salts are usually formed by intermolecular proton transfer, modifying the nature of the intermolecular interactions principally by allowing for charge-assisted hydrogen bonding.
A potential design criterion is to use the difference in pKa values of the molecular components to predict whether neutral or charged complexes will be formed in the solid-state.19–21 One commonly-applied criterion is that a ΔpKa of >3 predicts proton transfer and the formation of an ionic salt, whereas within the ΔpKa range of 0–3 it is not possible to predict whether proton transfer will take place.22 This so-called salt-cocrystal continuum poses a difficulty to crystal-engineering approaches targeting specific supramolecular assembly, but also represents an opportunity to explore uncharted territory. Operating within this region can potentially allow access to either charged or neutral states, which, given the typically very different physical properties of ionic salts and neutral cocrystals, presents opportunities for property tuning.
In general, within the salt-cocrystal continuum a molecular complex of given stoichiometry will adopt a single protonation state under all conditions. There are examples of protonation state varying with stoichiometry,22–25 but it is uncommon for multiple protonation states to be accessible under ambient conditions for the same stoichiometry. There is a potential analogy here with concomitant polymorphism, i.e. the simultaneous co-production of multiple solid forms with differing physical properties, but concomitant growth of charged and neutral forms of the same complex has not previously been observed.
In principle, the co-existence of charged and neutral forms presents an opportunity for property switching through solid-state proton transfer. Temperature-induced proton transfer in multicomponent organic crystals is known, but comparatively rare.26 Where it does occur, it can lead to properties including thermochromism,27–29 mixed thermo- and photochromism30,31 and ferroelectricity.21 For example, the thermochromic 2-iodoaniline/picric acid system29 crystallises in yellow and green ionic forms under ambient conditions, both of which convert to red forms when heated above 60 °C through proton transfer to the acid yielding a neutral charge-transfer complex. This system inspired us to explore this phenomenon as a general design strategy to obtain thermochromic molecular complexes utilising proton transfer, the like of which is not currently available.
In this article, we present a systematic study of molecular proton-transfer thermochromics based on combining 4-haloanilines (4-XA) and 4-halo-2-methylanilines (4-X-2-MA) with 3,5-dinitrobenzoic acid (3,5-DNBA). We selected these components because of the small pKa difference between them, which places them in the salt-cocrystal continuum. Previous studies have related proton transfer to thermochromism and photochromism in molecular27–30 and coordination-polymer31 systems, whereas here additional novelty is offered by the rare observation of coexisting metastable neutral and charged species under ambient conditions. There are only a few recent reports of salt and cocrystal forms being obtained under the same crystallisation conditions, with the two forms leading to different physical properties.33,34 Combining the aniline and acid components produces four new functional thermochromic systems whose formation deliberately utilises the ambiguity of the salt-cocrystal continuum, enabling the stability and temperature of transformation to be modified by selecting components within a specific ΔpKa range.
Experimental
Co-crystallisation
Co-crystallisations were attempted under a variety of conditions to obtain a series of 1:1 molecular complexes of 3,5-dinitrobenzoic acid (3,5-DNBA) with one of five haloaniline molecules, viz. 4-iodianiline (4-IA), 4-bromoaniline (4-BrA), 4-iodo-2-methylaniline (4-I-2-MA), 4-bromo-2-methylaniline (4-Br-2-MA) and 4-chloro-2-methylaniline (4-Cl-2-MA).
For most systems, the complexes were obtained under a range of crystallisation conditions, but not reproducibly, and obtaining single crystals in particular proved to be challenging. Repeated crystallisation trials often gave inconsistent results. Due to the metastability of the coloured neutral co-crystals, many of the batches were unstable under ambient conditions and spontaneously converted to the colourless salt forms, making preservation and characterisation difficult. For the same reason, mixtures of the two forms were often obtained, either due to concomitant growth or spontaneous interconversion, further complicating the characterisation.
The procedures below summarise the optimised crystallisation conditions for the ten complexes analysed in this work, with a full description provided as ESI.†
:1 complexes (1–3) and a methanol solvate (CCDC: YIVHES).35 Complex 1 was synthesised by slow evaporation of an ethanol solution at ambient temperature, yielding colourless needles. The synthesis of 2 has been reported previously (CCDC: YIVHAO).323 was grown from ethyl acetate at 40 °C, yielding small red block crystals.
:1 complexes 4 and 5. 4 was grown by slow evaporation of an ethanol solution at room temperature, yielding colourless needles. 5 was grown by slow evaporation of an acetonitrile solution at 40 °C, yielding yellow/orange block crystals.
:1 complexes of 4-I-2-MA 3,5-DNBA 6 and 7 were obtained as colourless and red block crystals, respectively. The synthesis of both has been reported previously (CCDC: YIVHUI, YIVHUI01).35
:1 complexes of 4-Br-2-MA and 3,5-DNBA 8 and 9 were obtained. Molecular complex 8 was grown by slow evaporation of a methanol solution at ambient temperature, yielding colourless block crystals. 9 was grown by slow evaporation of a methanol solution at 40 °C, yielding orange block crystals.
Crystallography
Single crystal X-ray diffraction data for 1 and 5 were collected at 100 K using a Rigaku Oxford Diffraction Gemini A Ultra diffractometer (Mo Kα radiation). Single crystal X-ray diffraction data for 3, 8, 9 and 10 were collected at 150 K using the same instrument. Single crystal X-ray diffraction data for 4 were collected at 150 K using a Rigaku Oxford Diffraction SuperNova diffractometer (Mo Kα radiation). The structures were solved by direct methods using SHELXS-97 or SHELXS-2013 and refined using SHELXL-2014,36 both within the WinGX program suite.37 Crystallographic data tables and further details of the structure refinement are given in the ESI.†Hot-stage microscopy
Hot-stage microscopy (HSM) analysis was conducted using a Mettler Toledo FP82 hot stage equipped with a Leica DM1000 microscope. A single crystal of the sample was placed on a slide in the sample chamber and heated and cooled between 30 °C and an end temperature approximately 10 °C above the phase-transition temperature at a ramp rate of 5 °C per minute, with the temperature program controlled by an FP90 central processor.Thermal analysis
Differential-scanning calorimetry (DSC) experiments were conducted using a TA Instruments Q20 differential-scanning calorimeter. Samples were ground very gently and 1–3 mg weighed into a Tzero™ aluminium pan and sealed with a Tzero™ aluminium lid using a press. An empty, sealed pan was used as the reference. Samples were heated and cooled in cycles at a rate of 5 °C per minute.Spectroscopy
UV-vis absorption spectra were collected using a Hamamatsu UV-vis light source (tungsten-halogen lamp) and J&M Analytik TIDAS 1 spectrometer. A single crystal of the sample was cut into a thin plate with approximate dimensions 0.3 × 0.3 × 0.1 mm. Standard data collections were conducted at ambient temperature using the Bio-Kine32 software, with absorbance measured in the range 350–800 nm. Thirty spectra were collected and averaged for each sample measurement.Computational modelling
Density-functional theory (DFT) calculations were carried out on the X-ray structures of 1–10 using the VASP code.38Calculations were performed using the PBEsol39 functional with the DFT-D3 dispersion correction40 (i.e. PBEsol + D3). A plane-wave basis-set cutoff of 850 eV was used in all calculations, and the electronic Brillouin zones were sampled using Γ-centred Monkhorst-Pack k-point meshes41 with subdivisions of 2 × 1 × 1 (1, 2), 3 × 3 × 1 (3), 3 × 1 × 1 (4, 5), 1 × 3 × 1 (6), 2 × 2 × 1 (7, 9) and 1 × 2 × 1 (8, 10). Projector augmented-wave (PAW) pseudopotentials42,43 were employed including the outermost s and p electrons in the valence region. Each structure was fully optimised, with tolerances of 10−8 eV and 10−2 eV Å−1 on the total energy and forces applied during the electronic wavefunction minimisation and geometry optimisation respectively.
Single-point electronic-structure calculations with the PBE0 hybrid functional44 were performed on optimised structures to obtain more accurate orbital energies and to evaluate the dielectric properties using the linear-optics routines in VASP.45 For these calculations, the number of electronic bands was increased to around 3× the default to converge the sum over unoccupied (virtual) states.
Further details of the computational modelling, including a comparison of PBEsol and PBEsol + D3 and a comparison of the optimised lattice parameters to the experimental X-ray structures, are provided in the ESI.†
Results and discussion
The ten 1:1 molecular complexes obtained by evaporative crystallization are summarised in Fig. 1 and Table 1. These include two polymorphs of one salt and four thermochromic pairs comprising colourless salts and neutral co-crystals. The neutral coloured forms of the 2-IA, 2-BrA, 4-I-2-MA and 4-Br-2-MA complexes all undergo thermochromic phase transitions to colourless salts, which are clearly visible by hot-stage microscopy (Fig. 2). In all cases the colour change occurs over a temperature range of width ∼7–22 °C, appearing as a visible wave propagating across the crystals from a corner.
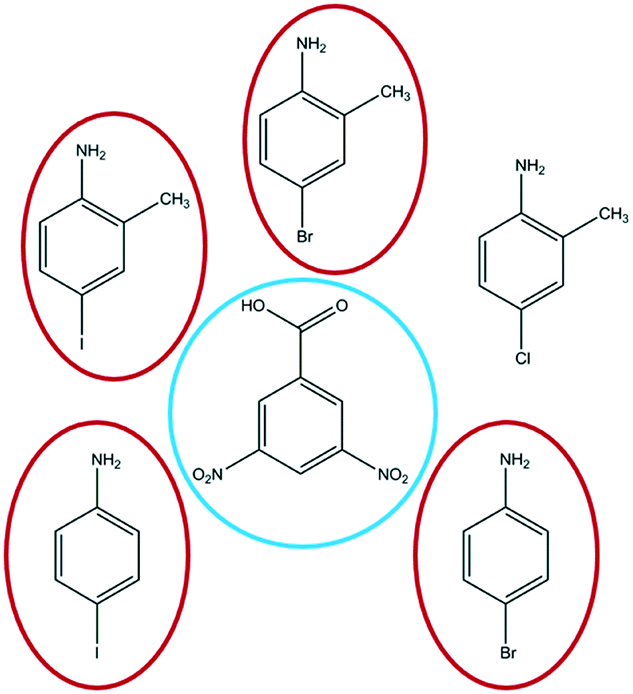 | ||
| Fig. 1 Molecular components employed in the co-crystallisation study in this work. 1:1 complexes were formed between the outer-ring haloaniline molecules and central 3,5-dinitrobenzoic acid. All molecules are shown in neutral form. Thermochromic pairs of neutral cocrystals and ionic salts were obtained with the four anilines circled in red, while only a colourless salt was formed with 4-chloro-2-methylaniline. | ||
| Molecular components | ΔpKa | Neutral (N) or ionic (I) | Conversion T (°C) | ΔEF (kJ mol−1 per F.U.) | Complex number |
|---|---|---|---|---|---|
| a Energy differences given for the two ionic polymorphs of the 4-IA system. | |||||
| 4-IA 3,5-DNBA | 0.99 | I, I, N | 98–108 | 11.86, 13.38a | 1, 2,323 |
| 4-BrA 3,5-DNBA | 1.07 | I, N | Unknown | 11.33 | 4, 5 |
| 4-I-2-MA 3,5-DNBA | 0.84 | I, N | 77–84 | 10.26 | 6,327 (ref. 32) |
| 4-Br-2-MA 3,5-DNBA | 0.89 | I, N | 61–83 | 8.51 | 8, 9 |
| 4-Cl-2-MA 3,5-DNBA | 1.03 | I | — | — | 10 |
 | ||
| Fig. 2 Thermochromic behavior of 3, 7 and 9 followed using hot-stage microscopy, showing the wave-like colour transformation across the crystal. No coloured complex was obtained for the 4-chloro-2-methylaniline system (10). | ||
3,5-DNBA is a light-yellow solid while 4-IA and 4-BrA are light-purple and beige, respectively, and the methyl-substituted anilines 4-I-2-MA/4-Cl-2-MA and 4-Br-2-MA are light purple and light brown in the solid state. Dissolution of 3,5-DNBA with the halogen components results in solutions ranging from yellow to orange and pale pink in colour. The strong colour of the neutral complexes therefore cannot be attributed solely to either component and most likely arises from charge-transfer processes.31,46–52
The pKa differences between the five anilines and 3,5-DNBA are all around 1 (Table 1), placing them firmly within the salt-cocrystal continuum. The most stable and easily-formed complexes are the colourless salts 1, 2,35,464, 6,35,468 and 10, whose crystal structures show 3D hydrogen bonding networks with no evidence for the formation of charge-transfer complexes (Fig. 3).
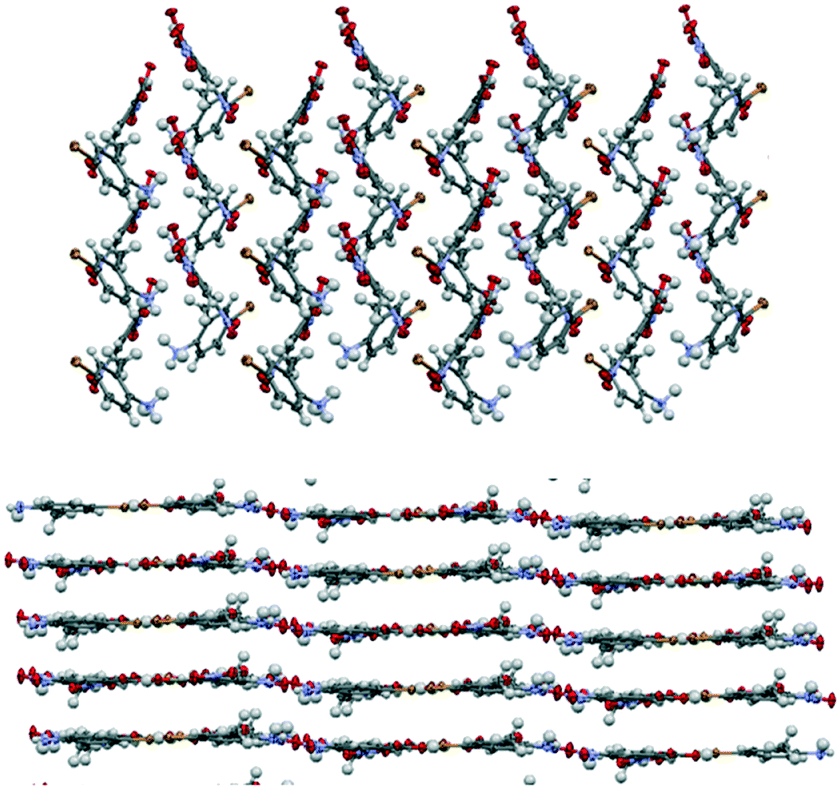 | ||
| Fig. 3 Typical crystal packing in the colourless salts (top) and coloured cocrystals (bottom) of the thermochromic molecular complexes. The three-dimensional hydrogen bonding in the ionic complexes results in a non-layered packing arrangement, whereas all of the neutral red/orange complexes adopt layered structures. These images show the 4-Br-2-MA complexes 8 and 9. Oxygen atoms are shown in red, iodine as purple, nitrogen as blue, carbon as grey and hydrogen as white. | ||
Uniquely, coloured neutral cocrystals of the iodo- and bromo-anilines (3, 5, 7 (ref. 35 and 46) and 9) were also isolated under ambient conditions, often co-existing with the colourless salts. Although these coloured neutral forms tended to be metastable, we were able both to obtain their crystal structures and to characterize their thermal behaviour. In contrast to the salts, these systems adopt layered structures, confirming the origin of the strong visible colour to be the formation of charge-transfer complexes. Fig. 3 compares the typical crystal packing in the salts and neutral cocrystals.
All six salts have a 1:1 stoichiometry and show full proton transfer from the carboxylic acid group of the acid to the amine group of aniline. 1, 2 and 4 crystallise in the orthorhombic space group P212121, whereas 6,358 and 10 crystallise in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) . Molecular complexes 1 and 4 are isomorphous (Fig. 4a), whereas complex 2 adopts a different structure (Fig. 4b) which is similar to the structure adopted by the isomorphous 6, 8 and 10 (Fig. 4c). The asymmetric units of 1 and 4 contain two independent 4-iodoanilinium cations and two independent 3,5-dinitrobenzoate anions, whereas the asymmetric unit of the other four salts contain one molecule each of the anion and cation.
. Molecular complexes 1 and 4 are isomorphous (Fig. 4a), whereas complex 2 adopts a different structure (Fig. 4b) which is similar to the structure adopted by the isomorphous 6, 8 and 10 (Fig. 4c). The asymmetric units of 1 and 4 contain two independent 4-iodoanilinium cations and two independent 3,5-dinitrobenzoate anions, whereas the asymmetric unit of the other four salts contain one molecule each of the anion and cation.
 | ||
| Fig. 4 Crystal packing in the ionic salts 1 and 4 (a), 2 (b) and 7, 8 and 10 (c). In the structure in (a) the independent 3,5-dinitrobenzoic acid molecules are colored yellow and red and the haloaniline molecules are blue and green. In (b) and (c), the acid and aniline components are coloured yellow and blue, respectively. | ||
In all six molecular complexes charge-assisted hydrogen bonding directs a three-dimensional network in which hydrogen bonding between the NH3+ groups of the cations and the oxygen atoms of the carboxylate groups of the 3,5-dinitrobenzoate anions result in the formation of R34(10) hydrogen-bonded rings (Fig. 5a).
 | ||
| Fig. 5 (a and b) Intermolecular interactions in 1 and 4. (a) Moderate strength N–H⋯O hydrogen bonds linking the two 4-XA cations and four 3,5-DNBA anions. (b) Hydrogen-bonded chain with a backbone of stacked 4-XA cations connecting adjacent chains via short C–X⋯O contacts. (c) Contrasting stacking in 2. Oxygen atoms are shown in red, iodine in purple, nitrogen in blue, carbon in grey and hydrogen in white, and the intermolecular contacts referred to in the discussion are indicated by blue dotted lines. | ||
In 1, 2 and 4, these four-molecule units extend along the crystallographic a-axis through two further hydrogen bonds to form a continuous hydrogen-bonded chain. In 1 and 4, a stack of parallel 4-haloanilinium cations form the backbone of the chain (Fig. 5b), and the halogen atom of one cation forms a short contact with one of the nitro group oxygen atoms in an adjacent 3,5-DNBA chain with I⋯O = 3.312(3) and Br⋯O = 3.184(4) Å, which can be compared to the sums of the van der Waals radii of 3.50 and 3.37 Å, respectively.
In complex 2 there is no stacking of the 4-IA molecules and the cations are oriented away from one other and on opposite sides of the chain (Fig. 5c). The iodine atoms form halogen bonds with one of the carboxylate oxygen atoms with an I⋯O distance of 2.987(2) Å, which is significantly shorter than the sum of the van der Waals radii (3.5 Å) but weaker than the halogen bond in 1. This interaction links the individual hydrogen-bonded chains along the direction of the crystallographic b-axis.
All three complexes also show Onitro⋯Onitro and Onitro⋯π interactions between 3,5-DNBA anions. Overall, the three-dimensional hydrogen bonding arrangement results in non-layered crystal structures, and there are no π⋯π stacking interactions between 4-XA and 3,5-DNBA molecules. Projection down the crystallographic b-axis shows the alternating sections of 4-XA and 3,5-DNBA molecules (Fig. 4a and b).
The 4-X-2-MA complexes show a similar set of intermolecular interactions. The methyl group is involved in one weak hydrogen bond to one of the carboxylate O atoms. The halogen atom of the 4-X-2-MA cation forms a short X⋯O contact to one of the nitro groups with I⋯O = 3.204(2), Br⋯O = 3.160(3) and Cl⋯O = 3.170(3) Å compared to the sum of the van der Waals radii of the two atoms of 3.50, 3.37 and 3.27 Å, respectively. In addition, there are also Onitro⋯Onitro interactions between adjacent 3,5-DNBA anions and O⋯π interactions involving nitro and carboxylate O atoms.
While not isomorphous, the four neutral cocrystals all adopt layered structures with alternate columnar stacking of the two components and similar intermolecular interactions. All four complexes crystallise in P. Complexes 3, 7 and 9 have one of each molecule in the asymmetric unit, while complex 5 has two.
In all four structures dimers are formed between equivalent coplanar 3,5-DNBA molecules through moderate strength O–H⋯O hydrogen bonds, producing the R22(8) rings typical for carboxylic acid groups, and alternating rows of the aniline molecules and acid dimers interact to form planar two-dimensional sheets. However, the intermolecular interactions and orientations of the aniline molecules within the chains differ between the complexes (Fig. 6).
 | ||
| Fig. 6 Intermolecular interactions in the two-dimensional sheets formed from hydrogen-bonded 3,5-DNBA dimers and aniline molecules in complexes 3 (a), 5 (b), 7 (c) and 9 (d). Oxygen atoms are shown in red, iodine in purple, nitrogen in blue, carbon in grey and hydrogen in white, and the intermolecular contacts referred to in the discussion are indicated by blue dotted lines. | ||
In complex 3, pairs of 4-IA molecules are in opposite orientations and the 3,5-DNBA dimers interact with one another through weak C–H⋯O hydrogen bonds to form a hydrogen-bonded chain (Fig. 6a). The 4-IA molecules form moderate-to-weak N–H⋯O hydrogen bonds to the nitro groups of the 3,5-DNBA molecules resulting in sheets. The iodine atoms do not participate in halogen interactions.
On the other hand, in 5 the 4-BrA molecules are in similar orientations across the chain. Each sheet incorporates both independent 4-BrA molecules and both 3,5-DNBA molecules in the asymmetric unit. Unlike 3, there are no interactions between adjacent 3,5-DNBA dimers. The H atoms of the amine groups on both 4-BrA molecules are involved in moderate to weak N–H⋯O hydrogen bonds with nitro group O atoms (Fig. 6b). In addition, one Br atom participates in a C–Br⋯O halogen bond with the nitro group oxygen atom with Br⋯O = 3.261(1) Å, compared to the sum of the van der Waals radii of Br and O of 3.37 Å. There are also further weak C–H⋯O hydrogen bonds between the aromatic hydrogen atoms of the 4-BrA molecules and the nitro/carboxylic acid group oxygen atoms (C⋯O = ∼3.3–3.5 Å).
In 7, the dimers interact with one another through weak C–H⋯O hydrogen bonds (C⋯O = 3.614(2) Å) to form the same hydrogen-bonded chain seen in 3 (Fig. 6c), whereas 9 adopts a different configuration in which rotation of the aniline molecule results in the 3,5-DNBA dimer units being pushed apart (Fig. 6d). As in the 4-XA-2-MA salts, the methyl group is again involved in only one weak hydrogen bond to a nitro group O atom, and the iodine atom is not involved in any interactions within the sheets.
The structure of 9 shows no interactions between the 3,5-DNBA dimers, with weak N–H⋯O hydrogen bonds between only one of the amine group H atoms and the nitro group oxygen atom. The methyl group is not involved in any interactions within the sheet, but there are interactions between the halogen and a nitro group O atom with Br⋯O = 3.120(2) Å, compared to the sum of the van der Waals radii of 3.37 Å.
In all four complexes, the hydrogen-bonding interactions between the 4-XA molecules and the 3,5-DNBA dimers within the sheets are weak or on the borderline between moderate and weak, and the distances are close to the sum of the van der Waals radii for hydrogen bonds. This highlights the poor H-bond donating ability of the NH2 groups and may explain why the neutral complexes are less stable than the salts. In general, however, the hydrogen bonds in the iodo complexes 3 and 7 are slightly stronger than those in the bromo complexes 5 and 9 respectively.
The planar sheets stack to form a layered structure, with alternately stacked 4-XA (electron-donor) and 3,5-DNBA (electron-acceptor) molecules forming mixed stack columns, and it is these aromatic donor⋯acceptor interactions that dominate the packing arrangement in the structures. In the 4-I-2-MA complex 7 there are aromatic donor⋯acceptor interactions between pairs of 4-I-2-MA and 3,5-DNBA molecules and π⋯π interactions between offset 3,5-DNBA molecules in adjacent layers. In the 4-Br-2-MA complex 9, aromatic donor⋯acceptor interactions are formed continuously along the stack but there are no offset π⋯π interactions between 3,5-DNBA molecules. 3 and 5 show very similar intermolecular separations of ∼3.36 and ∼3.32 Å between the stacked 4-XA and 3,5-DNBA molecules, as do the 4-X-2-MA analogues 7 and 9 at ∼3.23 and ∼3.30 Å. This stacking arrangement would support π⋯π* charge-transfer between the neutral aromatic donor and acceptor molecules in the mixed stacks.
Formation energies of 1–10 (ref. 53) calculated using density-functional theory (DFT) indicate the charged salt forms to be consistently more stable by ∼10 kJ mol−1 per formula unit than the corresponding neutral cocrystals (Table 1). This is in the range of typical energy differences between polymorphs and strengthens the analogy with the more “conventional” polymorphism observed in molecular solids. The colourless polymorphs of the 4-IA 3,5-DNBA salt (1, 2) form concomitantly, but 1 occurs in greater quantity and is formed more reliably. The higher density of 1 together with evidence from thermal analysis for the conversion of 2 to 1 at ∼98 °C prior to the melting of 1 at ∼115–116 °C (see ESI†) suggests higher stability.
The formation energies indicate a general stability order of I > Br > Cl (see ESI†), which is consistent with experimental transition temperatures of the Br and I systems (Table 1). On the other hand, the energy differences between the salts and cocrystals show the reverse trend, with higher phase-transition temperatures corresponding to larger energy differences and, therefore, a stronger energetic driving force for the transition. This suggests that kinetics play an important role in determining the transition temperature. Methyl substitution of the haloaniline increases the stability of both the salt and cocrystal forms relative to the molecular components in the gas phase.
Upon exposure to the atmosphere the metastable coloured cocrystals convert over time to the more stable colourless salts. Together with the calculated formation energies, this indicates that the neutral (coloured) forms are kinetically trapped during crystallization, allowing them to exist concomitantly with the charged colourless forms but rendering them susceptible to temperature-induced phase transitions.
The significant structural changes associated with the proton transfer lead to loss of the integrity of the crystal during the transformation, but the shape of the crystal is maintained, suggesting the process is not a recrystallisation. The temperature-induced conversion between neutral and ionic species could also be followed by powder X-ray diffraction (see ESI†).
The modelling studies show that the highest-occupied and lowest-unoccupied crystal orbitals (HOCOs) lie on the haloaniline and acid, respectively, in all four complexes (Fig. 7). This confirms that the optical absorption and reflected colour indeed arise from charge-transfer processes, and the calculated HOCO–LUCO gaps and simulated absorption spectra are both consistent with the bold colours of the neutral cocrystals. Following the proton transfer in the salts, the HOCO is lowered and the LUCO raised in energy, widening the gap and blue-shifting the onset of absorption (Fig. 8).
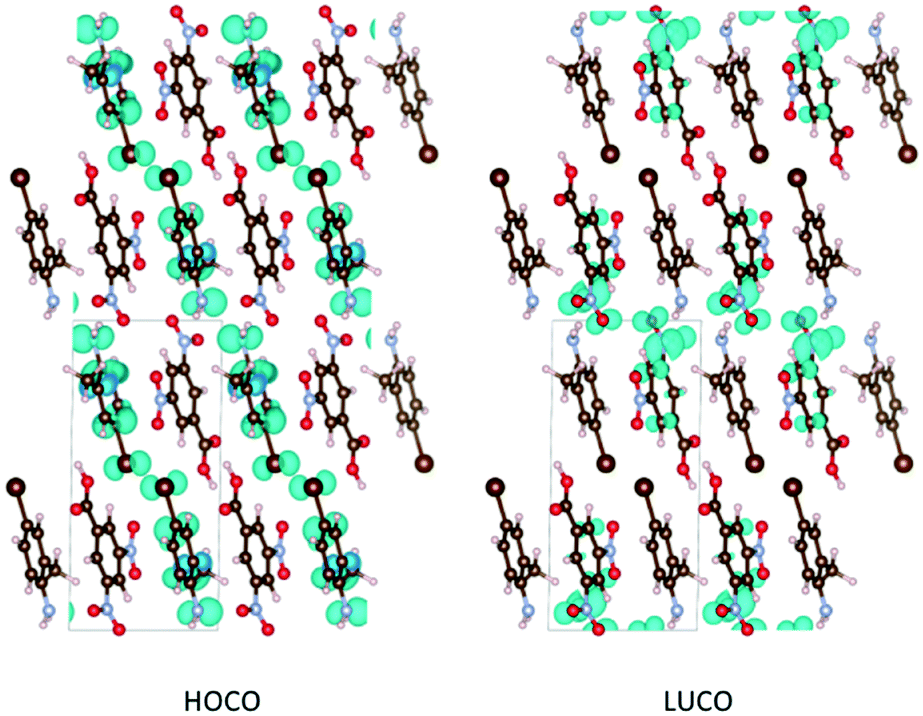 | ||
| Fig. 7 Orbital-density plots for the neutral complex 9 showing the aniline-based highest-occupied crystal orbital (HOCO) and acid–based lowest-unoccupied crystal orbital (LUCO). The lowest-lying optical transition in the simulated spectrum corresponds to a charge-transfer absorption between these states. | ||
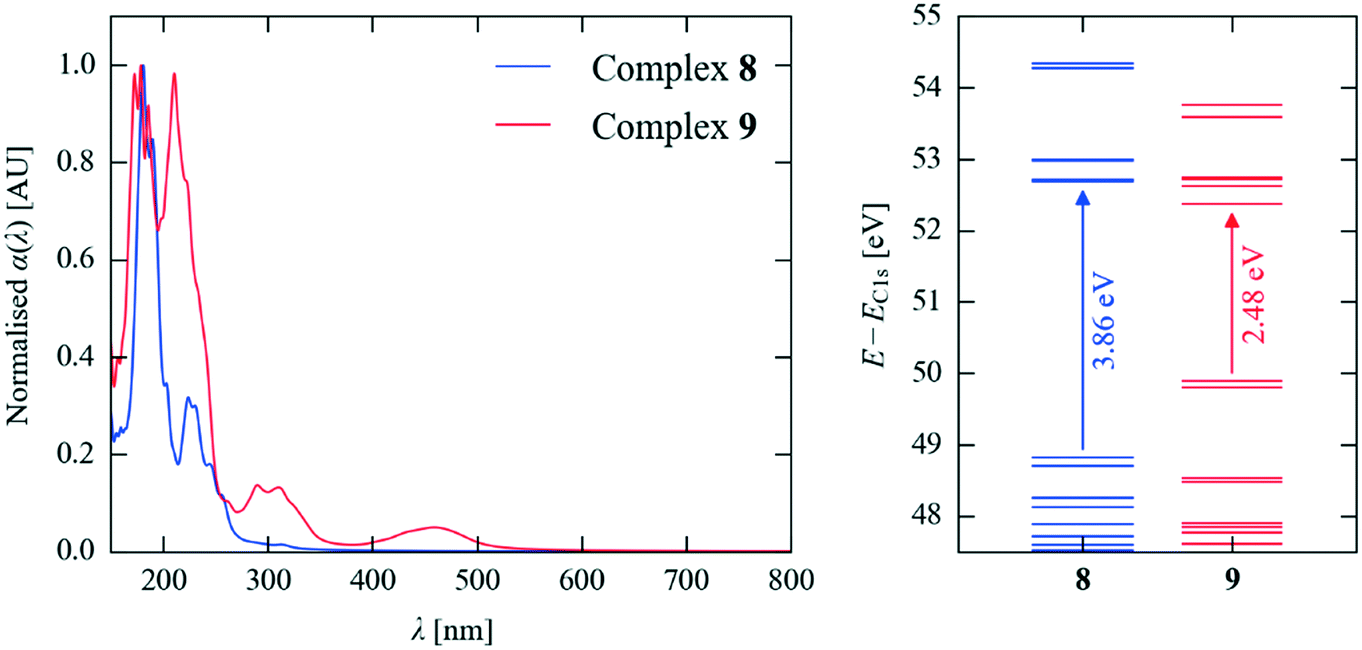 | ||
| Fig. 8 Simulated optical absorption spectra and energy-level spectra of the ionic complex 8 and neutral complex 9 showing the wider band gap and blue-shifted absorption spectrum of the salt. | ||
The colour of the cocrystals is tunable over a small range by varying the halogen atoms: the iodoanilines tend to give red crystals, whereas the bromoaniline cocrystals are orange. From the crystallography, the layer spacing of the two components in both derivatives is similar, and therefore the subtle differences in color must arise either from differences in molecular properties (linked directly to the halogen atom), differing relative orientations of the molecules in the charge-transfer stacks, and/or differences in hydrogen-bond strengths. The haloaniline-based HOCOs include a substantial contribution from the halogen atoms, so the higher-energy iodine lone pairs would be expected to raise the HOCO energy and produce a narrower HOCO–LUCO gap, stronger visible absorption and a red-shifted reflected color. This is borne out in the simulated spectra (see ESI†).
The introduction of the electron-donating methyl substituent leads to an ∼20 nm shift of the absorbance to longer wavelength, which can be attributed to a small influence on the crystal packing, altering the molecular overlap, and changes in the electron-donating properties of the aniline component.
Conclusions
In conclusion, four pairs of thermochromic complexes of haloanilines with 3,5-dinitrobenzoic acid have been isolated that, uniquely, exist both as salts and neutral cocrystals under ambient conditions. The transition temperatures can be tuned by the substituent chemistry, within an overall mechanism where the phase transitions and striking accompanying changes in colour are governed by proton transfer and associated structural rearrangements.The existence of both colourless ionic salts and coloured cocrystals can be partly explained by the ΔpKa values, all of which lie around 1, in a region where established literature precedents indicate that there is no clear preference between the two forms.
The calculated formation-energy differences between the neutral and ionic forms lie in a similar range to those found for conventional polymorphism. The coloured, neutral cocrystals are metastable, kinetically trapped during the crystallization process, and revert to the more stable (colourless) salt form over time or on heating.
The stability and colour of the metastable state is linked to the nature of the halogen substituent and can be further tuned by the addition of methyl substituents to the aniline ring. The crystal structures of the salts show a 3-dimensional hydrogen bonding network, whereas the cocrystals adopt layered structures in which π⋯π interactions facilitate charge transfer between aniline- and acid–based orbitals.
The coloured cocrystals undergo thermochromic solid-state phase transitions to colourless salts between 61–108 °C. The red iodo complexes transform at higher temperatures than the orange bromo analogues, suggesting higher stability, though comparison to formation energies from modelling studies suggests that kinetics may play a significant role in determining the phase-transition temperatures.
Exploring the ambiguous region of the salt-cocrystal continuum and identifying similar “indecisive” molecular complexes may provide novel routes to switchable systems exhibiting large changes in physical properties. In the case of thermochromism, the ΔpKa criterion alongside that of selecting suitable electron-donating/electron-accepting π systems with a tendency to form charge-transfer stacks may provide a design strategy towards obtaining systems where the transition temperatures can be systematically controlled using established crystal-engineering principles. The proton-transfer paradigm and the link between stability, colour change and molecular composition thus offers a new design route to tunable thermochromics for a range of potential applications.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge support from the UK Engineering and Physical Sciences Research Council (EPSRC) (grant no. EP/K004956/1 and EP/P007821/1). JMS is grateful to the University of Manchester for the support of a Presidential Fellowship. Calculations were performed on the UK National Archer HPC facility, accessed through membership of the UK Materials Chemistry Consortium and funded by the EPSRC (EP/L000202), and on the SiSu supercomputer at the IT Center for Science (CSC), Finland, via the Partnership for Advanced Computing in Europe (PRACE) project 13DECI0317/IsoSwitch.Notes and references
- M. Irie, Chem. Rev., 2000, 100, 1685 CrossRef CAS PubMed.
- J. H. Day, Chem. Rev., 1963, 63, 65–80 CrossRef CAS.
- T. Yuan, M. Vazquez, A. N. Goldner, Y. Xu, R. Contrucci, M. A. Firestone, M. A. Olson and L. Fang, Adv. Funct. Mater., 2016, 26, 8604 CrossRef CAS.
- A. Seeboth, R. Ruhmann and O. Mühling, Materials, 2010, 3, 5143 CrossRef CAS PubMed.
- E. L. Harty, A. R. Ha, M. R. Warren, A. L. Thompson, D. R. Allan, A. L. Goodwin and N. P. Funnell, Chem. Commun., 2015, 51, 10608 RSC.
- C. B. Aakeröy, N. R. Champness and C. Janiak, CrystEngComm, 2010, 12, 22 RSC.
- O. Almarsson and M. J. Zaworotko, Chem. Commun., 2004, 1889 RSC.
- T. Friščić and W. Jones, J. Pharm. Pharmacol., 2010, 62, 1547–1559 CrossRef PubMed.
- P. Vishweshwar, J. A. McMahon, J. A. Bis and M. J. Zaworotko, J. Pharm. Sci., 2006, 95, 499 CrossRef CAS PubMed.
- F. Pan, M. S. Wong, V. Gramlich, C. Bosshard and P. Gu, J. Am. Chem. Soc., 1996, 118, 6315 CrossRef CAS.
- E.-Y. Choi, M. Jazbinsek, S.-H. Lee, P. Günter, H. Yun, S. W. Lee and O.-P. Kwon, CrystEngComm, 2012, 14, 4306 RSC.
- D. Yan, A. Delori, G. O. Lloyd, T. Friščić, G. M. Day, W. Jones, J. Lu, M. Wei, D. G. Evans and X. Duan, Angew. Chem., Int. Ed., 2011, 50, 12483 CrossRef CAS PubMed.
- D. Yan, A. Delori, G. O. Lloyd, B. Patel, T. Friščić, G. M. Day, D.-K. Bučar, W. Jones, J. Lu, M. Wei, D. G. Evans and X. Duan, CrystEngComm, 2012, 14, 5121 RSC.
- Z.-P. Deng, L.-H. Huo, H. Zhao and S. Gao, Cryst. Growth Des., 2012, 12, 3342 CrossRef CAS.
- Q. Feng, M. Wang, B. Dong, C. Xu, J. Zhao and H. Zhang, CrystEngComm, 2013, 15, 3623 RSC.
- C. A. Gunawardana and C. B. Aakeroy, Chem. Commun., 2018, 54, 14047 RSC.
- S. Mohamed, A. A. Alwan, T. Friscic, A. J. Morris and M. Arhangelskis, Faraday Discuss., 2018, 211, 401 RSC.
- A. O. Weldeab, A. Steen, D. J. Starkenburg, J. S. Dal Williams, K. A. Abboud, J. G. Xue, N. I. Hammer, R. K. Castellano and D. L. Watkins, J. Mater. Chem. C, 2018, 6, 11992 RSC.
- B. R. Bhogala, S. Basavoju and A. Nangia, CrystEngComm, 2005, 7, 551 RSC.
- K.-S. Huang, D. Britton, L. Margaret, T. C. Etter and S. R. Byrn, J. Mater. Chem., 1997, 7, 713 RSC.
- A. J. Cruz-Cabeza, CrystEngComm, 2012, 14, 6362 RSC.
- S. L. Childs, G. P. Stahly and A. Park, Mol. Pharmaceutics, 2007, 4, 323 CrossRef CAS PubMed.
- D. A. Haynes, W. Jones and W. D. S. Motherwell, CrystEngComm, 2006, 8, 830 RSC.
- D. A. Haynes and L. K. Pietersen, CrystEngComm, 2008, 10, 518 RSC.
- C. C. Seaton, T. Munshi, S. E. Williams and I. J. Scowen, CrystEngComm, 2013, 15, 5250 RSC.
- A. O. F. Jones, M.-H. Lemee-Cailleau, D. M. S. Martins, G. J. McIntyre, I. D. H. Oswald, C. R. Pulham, C. K. Spanswick, L. H. Thomas and C. C. Wilson, Phys. Chem. Chem. Phys., 2012, 14, 13273 RSC.
- D. M. S. Martins, D. S. Middlemiss, C. R. Pulham, C. C. Wilson, M. T. Weller, P. F. Henry, N. Shankland, K. Shankland, W. G. Marshall, R. M. Ibberson, K. Knight, S. Moggach, M. Brunelli and C. A. Morrison, J. Am. Chem. Soc., 2009, 131, 3884 CrossRef CAS PubMed.
- M. T. Reetz, S. Hoger and K. Harms, Angew. Chem., Int. Ed. Engl., 1994, 33, 181 CrossRef.
- M. Tanaka, H. Matsui, J. Mizoguchi and S. Kashino, Bull. Chem. Soc. Jpn., 1994, 67, 1572 CrossRef CAS.
- F. Robert, A. D. Naik, F. Hidara, B. Tinant, R. Robiette, J. Wouters and Y. Garcia, Eur. J. Org. Chem., 2010, 4, 621 CrossRef CAS; P.-L. Jacquemin, K. Robeyns, M. Devillers and Y. Garcia, Chem. – Eur. J., 2015, 21, 6832 CrossRef PubMed.
- T. Haneda, M. Kawano, T. Kojima and M. Fujita, Angew. Chem., Int. Ed., 2007, 46, 6643 CrossRef CAS PubMed.
- S. Horiuchi and Y. Tokura, Nat. Mater., 2008, 7, 357 CrossRef CAS PubMed.
- X. Fu, J. Li, L. Wang, B. Wu, X. Xu, Z. Deng and H. Zhang, RSC Adv., 2016, 6, 26474 RSC; E. A. Buvaylo, V. N. Kokozay, N. Yu. Strutynska, O. Yu. Vassilyeva and B. W. Skelton, Acta Crystallogr., Sect. C: Struct. Chem., 2018, 74, 152 CrossRef CAS PubMed.
- S. R. Perumalla, C. Wang, Y. Guo, L. Shi and C. C. Sun, CrystEngComm, 2019 10.1039/c8ce01076e.
- C. L. Jones, C. C. Wilson and L. H. Thomas, CrystEngComm, 2014, 16, 5849 RSC.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- L. J. Farrugia, J. Appl. Crystallogr., 2012, 45, 849–854 CrossRef CAS.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 558(R) CrossRef.
- J. P. Perdew, A. Ruzsinszky, G. I. Csonka, O. A. Vydrov, G. E. Scuseria, L. A. Constantin, X. Zhou and K. Burke, Phys. Rev. Lett., 2008, 100, 136406 CrossRef PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Solid State, 1976, 13, 5188 CrossRef.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953 CrossRef.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758 CrossRef CAS.
- C. Adamo, J. Chem. Phys., 1999, 110, 6158 CrossRef CAS.
- M. Gajdoš, K. Hummer, G. Kresse, J. Furthmüller and F. Bechstedt, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 73, 045112 CrossRef.
- S. Horiuchi and Y. Tokura, Nat. Mater., 2008, 7, 357 CrossRef CAS PubMed.
- R. S. Mulliken, J. Am. Chem. Soc., 1952, 74, 811 CrossRef CAS.
- K. P. Goetz, D. Vermeulen, M. E. Payne, C. Kloc, L. E. McNeil and O. D. Jurchescu, J. Mater. Chem. C, 2014, 2, 3065 RSC.
- A. Das and S. Ghosh, Angew. Chem., Int. Ed., 2014, 53, 2038 CrossRef CAS PubMed.
- C. C. Seaton, N. Blagden, T. Munshi and I. J. Scowen, Chem. – Eur. J., 2013, 19, 10663 CrossRef CAS PubMed.
- W. Zhu, R. Zheng, X. Fu, H. Fu, Q. Shi, Y. Zhen, H. Dong and W. Hu, Angew. Chem., Int. Ed., 2015, 54, 6785 CrossRef CAS PubMed.
- W. Zhu, R. Zheng, Y. Zhen, Z. Yu, H. Dong, H. Fu, Q. Shi and W. Hu, J. Am. Chem. Soc., 2015, 137, 11038 CrossRef CAS PubMed.
- A. Pallipurath, J. M. Skelton, A. Delori, C. Duffy, A. Erxleben and W. Jones, CrystEngComm, 2015, 17, 7684 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Includes further details of the synthesis and characterisation of complexes 1–10, crystallographic data including details of the structure refinement, additional spectroscopic data, and a full description and analysis of the computational modelling. CCDC structure deposition numbers: 1556544–1556550. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8ce02066c |
| This journal is © The Royal Society of Chemistry 2019 |