
Halogen and chalcogen-bonding interactions in sulphur-rich π-electron acceptors†
Yann
Le Gal
,
Adrien
Colas
,
Frédéric
Barrière
,
Vincent
Dorcet
,
Thierry
Roisnel
and
Dominique
Lorcy
 *
*
Univ Rennes, CNRS, ISCR (Institut des Sciences Chimiques de Rennes) - UMR 6226, F-35000 Rennes, France. E-mail: Dominique.lorcy@univ-rennes1.fr
First published on 20th February 2019
Abstract
In order to explore the feasibility of generating halogen bonding interactions between sulphur-rich π-electron acceptors, we prepared three bithiazolidinylidene derivatives substituted by iodine atoms, namely 3,3′-bis(iodophenyl)bithiazolidinylidene-2,4,2′,4′-tetrathione (BIP-BTTT). Sulphur and iodine heteroatoms were introduced to the skeleton of the acceptor molecule to induce chalcogen⋯chalcogen and halogen bonding interactions. Both interactions can be evidenced by X-ray diffraction studies in the synthetic precursors as well as in the acceptors themselves.
Introduction
The design of molecules for the elaboration of semi-conducting molecular materials relies essentially on the intrinsic properties of the molecular precursors. However, the properties of the materials are also based on the solid state organization and on the intermolecular interactions between molecules within the material.1,2 Indeed, weak interactions between molecules will result in an insulating material with no charge carrier delocalization. Various types of intermolecular interactions have been explored such as chalcogen⋯chalcogen contacts,3 hydrogen bonding,4,5 π-π stacking,6 and halogen bonding,7 either separately or cooperatively.8 In this context, we recently developed the synthesis of sulphur-rich electron acceptors with a bithiazolidinylidene-2,4,2′,4′-tetrathione (BTTT) backbone (Chart 1).9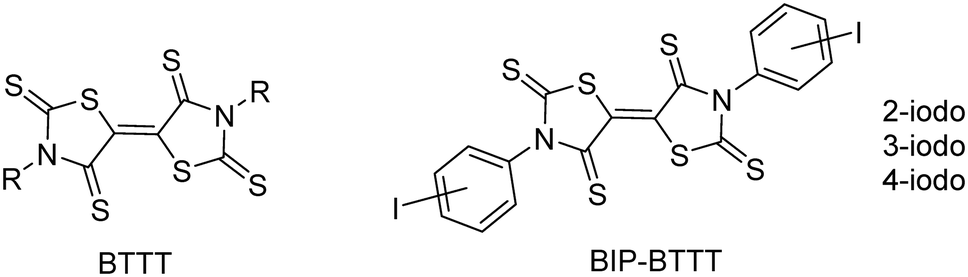 | ||
| Chart 1 | ||
Several members of this family led to air-stable n-channel organic field effect transistors (OFETs) exhibiting good performances.10,11 The air stability and the enhanced charge mobility were ascribed to the presence of S⋯S intermolecular interactions.10 As the intermolecular interactions are of high importance for the charge mobility as well as for the stability of n-channel OFETs,12 we focused our interest on interactions other than chalcogen⋯chalcogen contacts. For this purpose, we investigated the synthesis of similar acceptors substituted with halogen atoms such as iodine, potentially able to form halogen bonds (XBs) with the chalcogen atoms of the neighbouring molecules, acting as XB acceptors. Indeed, we recently reported for example that the exocyclic sulphur atom in thiazoline-2-thiones can act as an efficient XB acceptor toward organic iodinated molecules.13 Therefore we investigated the synthesis of 3,3′-bis(iodophenyl)-BTTT, BIP-BTTT (Chart 1) where the nitrogen atoms of the heterocycles, the thiazoline-2-thione moieties, are substituted by an ortho, meta or para iodophenyl moiety. These different substitution patterns led to the analysis of the influence of the localization of the halogen atom on the intermolecular interactions. In this study, we report the syntheses and X-ray structure investigations of the precursors and the novel electron acceptors BIP-BTTT. Electrostatic surface potential (ESP) calculations carried out on four of the crystallographically characterized compounds provide a complementary understanding of the organization of the molecules in the solid state.
Results and discussion
The strategy we used to synthesize the acceptors BIP-BTTT 4a–c starting from 2-, 3- and 4-iodoaniline is outlined in Scheme 1. Addition of triethylamine to a solution of the iodoaniline in carbon disulphide led to the dithiocarbamate salts 1a–c in good yields. Alkylation of 1a–c with chloroethanal followed by cyclization and dehydration in the presence of sulphuric acid allowed us to isolate the thiazoline-2-thiones 2a–c. The next step involves the construction of the fused dithiole-2-one rings on the thiazoline-2-thiones 2a–c. Successive addition of LDA and sulphur to thiazoline-2-thiones 2a–c followed by the addition of triphosgene afforded the dithiol-2-ones 3a–c. We previously demonstrated that a thermal treatment of such protected dithiolene ligands within a dithiol-2-one cycle is an efficient strategy for the synthesis of the acceptor skeleton.9 The acceptors BIP-BTTT 4a–c were therefore synthesized by simply heating the bicyclic structures 3a–c in refluxing toluene. These acceptors 4a–c were obtained as deep purple compounds poorly soluble in common organic solvents, the less soluble one being para-substituted 4a.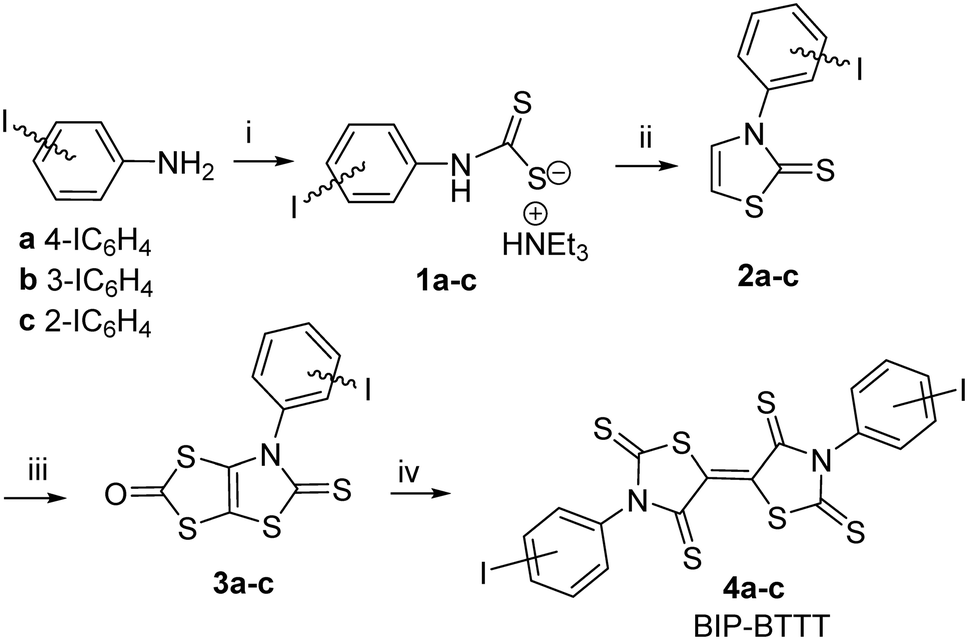 | ||
| Scheme 1 Synthetic path to BIP-BTTT derivatives 4a–c, 26–36% yield from 2a–c. Reactants and conditions: i) CS2, NEt3; ii) ClCH2CHO, H2SO4; iii) LDA, S8, (Cl3CO)2CO; iv) toluene, Δ. | ||
Electrochemical investigations carried out by cyclic voltammetry allowed us to determine the redox potentials of these derivatives. They were performed in DMSO for the three acceptors 4a–c and in CH2Cl2 only for 4b–c, because 4a is not soluble enough in dichloromethane. The redox potentials are collected in Table 1 together with those of DEBTTT (R = Et, Chart 1) for comparison. Two reversible monoelectronic reduction waves are observed for these three acceptors, either in DMSO or in CH2Cl2, attributed to the successive reduction of the acceptor into a radical anion and a dianion. Compared to DEBTTT, in both solvents the redox potentials of the acceptors 4a–c are slightly shifted towards more anodic potentials, indicating a weak effect of the iodophenyl substituent on the overall accepting ability of these molecules. 4a–c and DEBTTT exhibit slightly lower electron accepting ability than TCNQ (E1 = 0.18 V and E2 = −0.37 V vs. SCE).14
| Acceptors | CH2Cl2 | DMSO | ||
|---|---|---|---|---|
| E 1/2 1 | E 1/2 2 | E 1/2 1 | E 1/2 2 | |
| DEBTTT | −0.05 | −0.44 | 0.06 | −0.41 |
| 4a | 0.09 | −0.33 | ||
| 4b | −0.01 | −0.43 | 0.1 | −0.34 |
| 4c | −0.04 | −0.48 | 0.05 | −0.37 |
Crystals of sufficient quality for an X-ray diffraction study were obtained by slow concentration of a CH2Cl2 solution of 2b, 3a and 3c as well as from a CHCl3 solution of acceptor 4c. The molecular structure of 2b is reported in Fig. 1. The thiazole core is planar, while the phenyl ring is located in a plane forming a dihedral angle of 63.4(5)° with the thiazoline-2-thione core. The sulphur atoms can behave as a Lewis base and form intermolecular halogen bonding with the iodine atom of a neighbouring molecule.13,15 However in 2b, only a short distance between the hydrogen atom of the thiazoline ring and the sulphur atom of the thione (2.788(4) Å), assigned to a hydrogen bond (HB), was observed.
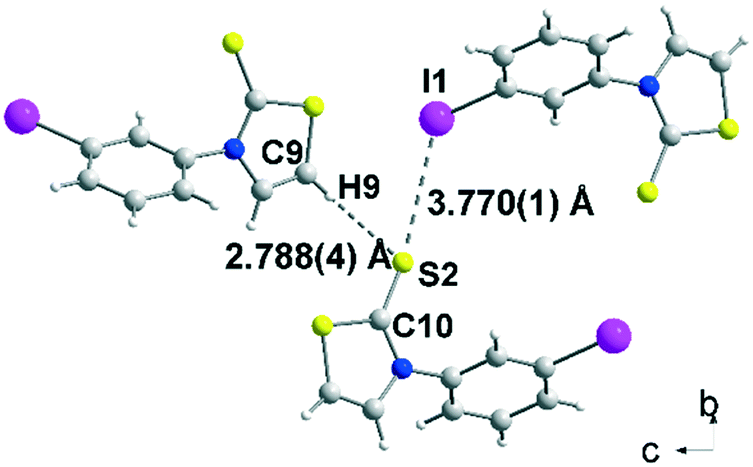 | ||
Fig. 1 View of the C–H⋯S![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) and C–I⋯S interactions on the bc plane for 2b. and C–I⋯S interactions on the bc plane for 2b. | ||
The molecular structures of dithiol-2-ones 3a and 3c are reported in Fig. 2. For both derivatives, the dithiol-2-one and the fused thiazoline core are coplanar and form with the plane of the aromatic substituent a dihedral angle of 50.5(2)° for 3a and 100.2(3)° for 3c. Short I⋯O contacts are observed between two neighbouring molecules of 3.271 Å for 3a and 3.252 Å for 3c, corresponding to a reduction ratio of 93.4% and 92.9% respectively, relative to the van der Waals contact distance (3.50 Å), indicating a sizeable XB interaction between neighbouring molecules (Fig. 2a and b).16 The C–I⋯O angles, at 161.4° and 154.6°, are here closer to linearity and consistent with an XB interaction. In addition, for both structures, chalcogen⋯chalcogen contacts are also observed between neighboring molecules, at a distance shorter than the sum of the van der Waals radii, either between two sulphur centers (3.60 Å) or between sulphur and oxygen atoms (3.32 Å). The shortest sulphur⋯sulphur contacts measured for 3a and 3c are reported in Fig. 2c and d. The crystal structure of 3a reveals S⋯O and S⋯S distances of 3.274(12) Å and 3.329(5) Å, respectively, the latter corresponds to 93% of the van der Waals distance and is due to strong noncovalent bonding between two molecules. Comparatively, the S⋯S distances within 3c (the shortest being 3.416(1) Å) are longer than those in 3a. This is ascribed to a steric effect in the solid state of the ortho position of the iodine atom on the phenyl ring.
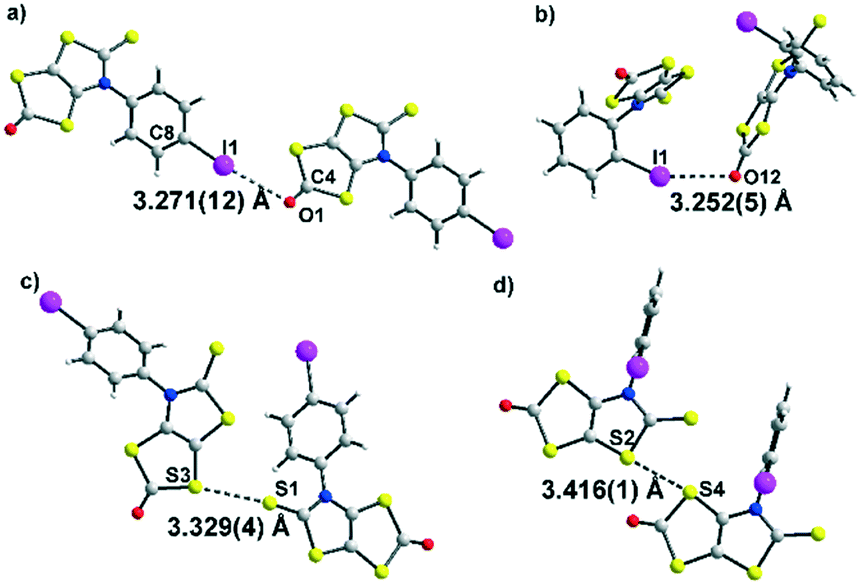 | ||
| Fig. 2 View of the XB interactions; O⋯I contacts shorter than the sum of the van der Waals radii in dark grey dotted lines in 3a (a) and 3c (b). View of the chalcogen interactions; S⋯S contacts shorter than the sum of the van der Waals radii in dark grey dotted lines in 3a (c) and 3c (d). | ||
Among the three electron acceptors, only the ortho substituted derivative 4c could be analysed by X-ray diffraction. The molecular structure of this derivative is presented in Fig. 3. It crystallizes in the triclinic system, space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , with two crystallographically independent molecules in the unit cell in general positions. This acceptor exhibits a planar skeleton and a trans configuration of the two thiazoline-2-thione rings with short intramolecular S⋯S contacts between the S atom of a thiocarbonyl group and the S atom of the thiazole ring (2.94 Å/2.91 Å). These short S⋯S contacts are in the same range as those observed for different acceptors belonging to the same family.9
, with two crystallographically independent molecules in the unit cell in general positions. This acceptor exhibits a planar skeleton and a trans configuration of the two thiazoline-2-thione rings with short intramolecular S⋯S contacts between the S atom of a thiocarbonyl group and the S atom of the thiazole ring (2.94 Å/2.91 Å). These short S⋯S contacts are in the same range as those observed for different acceptors belonging to the same family.9
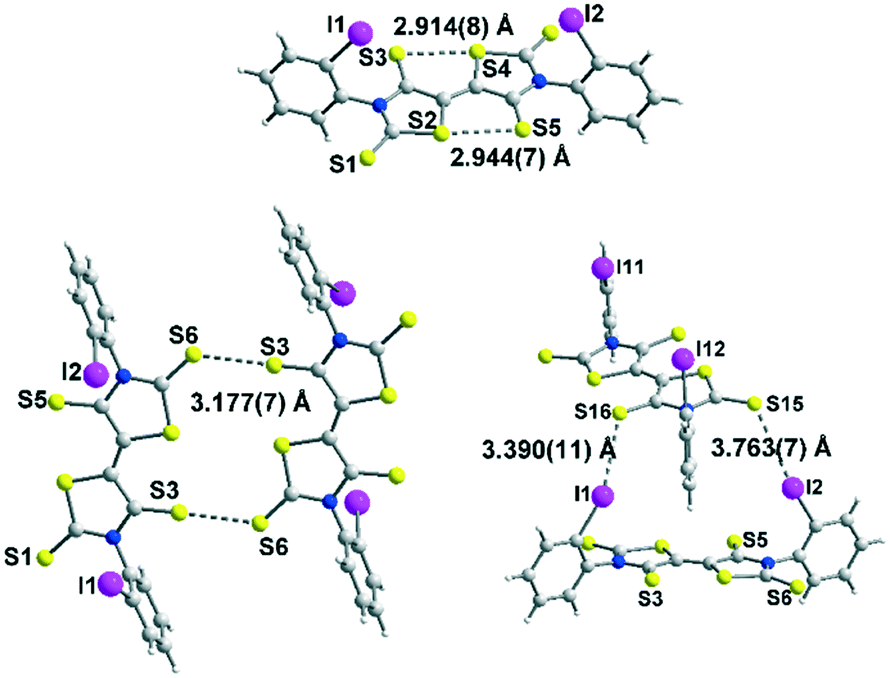 | ||
| Fig. 3 Molecular structure of acceptor 4c showing the intramolecular S⋯S contacts (top), the shortest intermolecular contacts (bottom left) and the XB bonds between two neighbouring molecules (bottom right). | ||
Interestingly, the ortho-iodophenyl substituents on the nitrogen atoms have a similar orientation of the iodine pointing in the same direction above the plane of the acceptor. The plane of the phenyl rings is almost perpendicular to the plane of the acceptor. The molecules are associated through I⋯SC XB interaction networks between I1⋯S16 3.390(4) Å and I11⋯S5 3.533(4) Å for the most significant short distances corresponding respectively to 89.7% and 93.4% of the van der Waals distances. Besides these intramolecular contacts, short S⋯S intermolecular contacts are also observed between neighbouring molecules, S3⋯S6 3.177(7) Å lower than the van der Waals radius of sulphur, corresponding to a reduction ratio of 88.2%. Nevertheless, due to steric hindrance generated by the iodophenyl rings, these S⋯S contacts within these acceptors are less numerous than those observed with DEBTTT where extensive three-dimensional S⋯S interactions were noticed.10
Electrostatic surface potential calculations have been performed on the optimized geometry of the four molecules that have been crystallographically characterized, namely 2b, 3a, 3c and 4c. These calculations were carried out in order to estimate the halogen bond donor abilities of the iodophenyl substituent within these different structures and to rationalize the interactions taking place in the crystals.9 As shown in Fig. 4, for 2b the maximum calculated positive electrostatic surface potential (ESP) is found at the hydrogen atoms located on the thiazole ring (+31.74 kcal mol−1) compared to only +23.89 kcal mol−1 at the iodine atom. The most negative calculated ESP is −29.09 kcal mol−1 and located on the thione's sulphur atom (CS). This calculated charge repartition is in good agreement with the organization of molecule 2b in the solid state where predominant hydrogen bonding interactions were observed between the hydrogen atom on the thiazoline ring and the sulphur atom of the thione, while no specific halogen bonding interaction involving the iodine atom was detected.
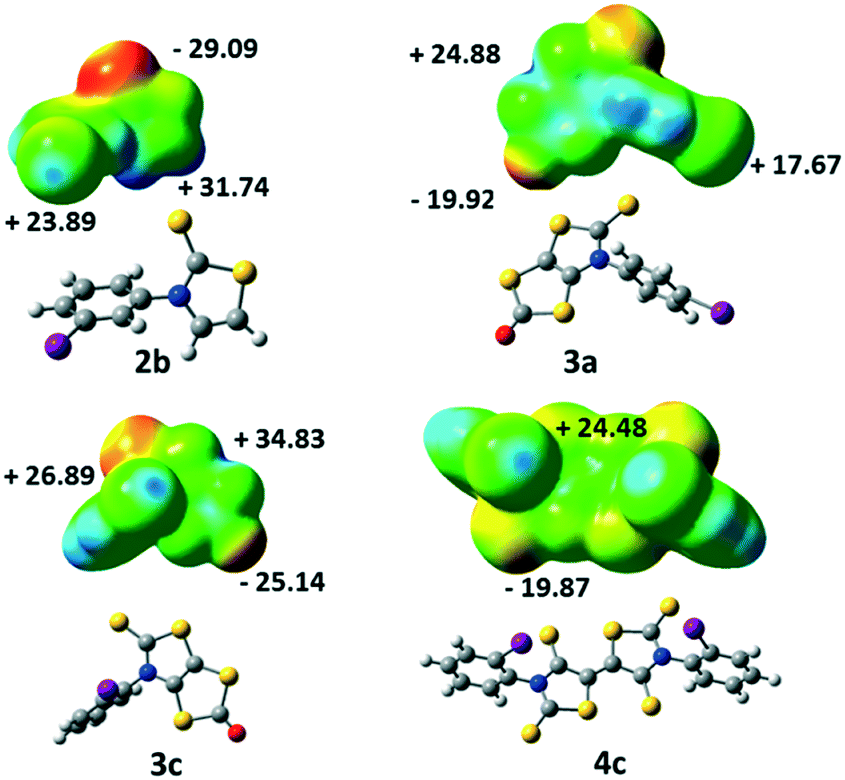 | ||
| Fig. 4 Molecular electrostatic potential surface mapped at the 0.001 e− au−3 isodensity surface for 2b, 3a, 3c and 4c. The common colour scale ranges from −29 kcal mol−1 (red) to +35 kcal mol−1 (blue). | ||
For the dithiol-2-ones 3a and 3c, the most negative calculated ESP is now located on the oxygen atom of the dithiole rings (CO, −19.92 and −25.14 kcal mol−1, respectively), while the most positive ESP is on the bicyclic structure (+24.88 and +34.83 kcal mol−1). The electrostatic surface potential value on the σ-hole of the halogen is lower (blue dot on the iodine atom in Fig. 4, +17.67 and +26.89 kcal mol−1). Despite these slightly lower values, the organization of both molecules in the solid state shows XB interactions (CO⋯I) between their iodine atom and the oxygen atom of a vicinal molecule.
Acceptor 4c is the only example where the highest ESP value over the whole molecule is found at the σ-hole of the iodine atom (+24.48 kcal mol−1, Fig. 4) while for the three other derivatives, it was either found on a hydrogen atom for 2b or on the bicycle structure for 3. The most negative calculated ESP for 4c is found on the sulphur atoms of the thione (−19.87 kcal mol−1). In accordance with these calculations, the iodine atom of this acceptor is indeed involved in short XB interactions with the thione's sulphur atom in the solid state (Table 2).
| Distances (Å) | Angles (°) | ||
|---|---|---|---|
| 2b | S2⋯H 2.788(4) | C9–H9⋯S2 175.9(3) | H9⋯S2C10 98.4(2) |
| 3a | O1⋯I1 3.271(12) | C8–I1⋯O1 161.4(4) | I1⋯O1C4 109.2(8) |
| S1⋯S3 3.329(4) | |||
| 3c | O12⋯I1 3.252(5) | C1–I1⋯O12 154.6(9) | I1⋯O12C11 111.1(2) |
| S2⋯S4 3.416(1) | |||
| 4c | S15⋯I2 3.763(7) | C21–I2⋯S15 155.5(5) | I2⋯S15C64 118.7(6) |
| S16⋯I1 3.390(4) | C1–I1⋯S16 158.2(3) | I1⋯S16C62 119.4(6) |
|
| S5⋯I11 3.533(4) | C51–I11⋯S5 156.3(3) | C12S5⋯I11 124.9(5) |
|
| S3⋯S6 3.177(7) | |||
We also performed some ESP calculations for the two other acceptors that have not been crystallographically characterized in order to analyse the influence of the localization of the halogen atom. For both acceptors, 4a and 4b, the most negative calculated ESP is located on the thione's sulphur atom (CS) as with acceptor 4c. On the other hand, the highest ESP value over the whole molecules 4a and 4b is not found at the σ-hole like for 4c (Fig. 5) but located para to the iodine atom on the aromatic ring, potentially a less favorable charge distribution for the growth of crystals in these cases.
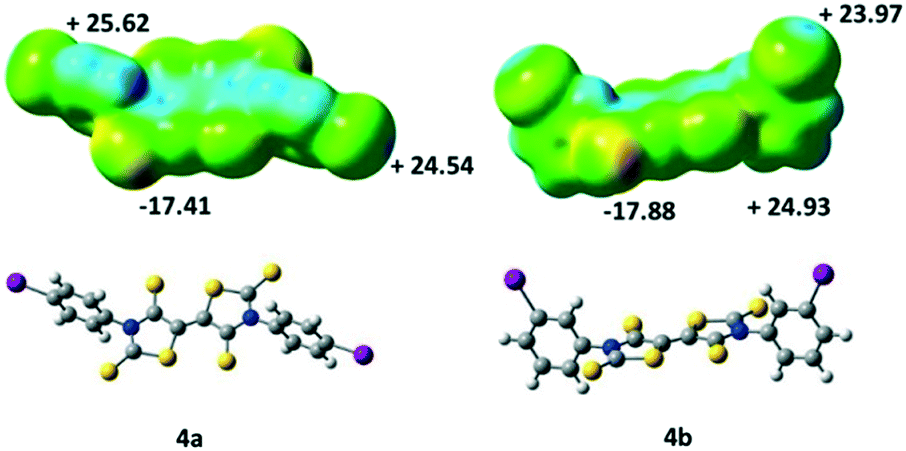 | ||
| Fig. 5 Molecular electrostatic potential surface mapped at the 0.001 e− au−3 isodensity surface for 4a and 4b. The colour scales range from −29 kcal mol−1 (red) to +35 kcal mol−1 (blue). | ||
Conclusions
In this study we investigated the synthesis of sulphur-rich electron π-acceptors, bithiazolidinylidenes, substituted by iodophenyl substituents, which can act as halogen bond donors toward thione (CS) or ketone (CO) groups acting as XB acceptors. Single crystals of the synthetic intermediates as well as one of the acceptors have been obtained. For the precursors, when the thione (CS) and ketone (CO) groups are present on the molecule, a halogen bond is formed between the iodine atom of one molecule and the CS/CO moieties of the neighbouring ones. For acceptor 4c, in accordance with the calculated ESP maxima, I⋯SC halogen bonds are observed between neighbouring molecules as well as S⋯S interactions. Thus we managed to observe from these BIP-BTTT acceptors the coexistence of two types of intermolecular interactions in the solid state. The next step will be the design of sulphur-rich electron acceptors substituted by groups bearing an iodine atom but less bulky than phenyl in order to increase the strength of the intermolecular interactions.
Experimental section
All commercial chemicals were used without further purification. The solvents were purified and dried by standard methods. All the NMR spectra were obtained in CDCl3 unless indicated otherwise. Chemical shifts are reported in ppm and 1H NMR spectra were referenced to residual CHCl3 (7.26 ppm) and 13C NMR spectra were referenced to CHCl3 (77.2 ppm). The 13C NMR spectra of acceptors 4a–c could not be obtained due to their low solubility. Melting points were measured on a Kofler hot-stage apparatus and are uncorrected. Mass spectrometry and elemental analyses were performed at the Centre Régional de Mesures Physiques de l'Ouest, Rennes. Cyclic voltammetry was carried out on a 10−3 M solution in CH2Cl2, containing 0.1 M nBu4NPF6 as the supporting electrolyte. Voltammograms were recorded at 0.1 Vs−1 on a platinum electrode and the potentials were measured versus the saturated calomel electrode (SCE).N-Iodophenyl-1,3-thiazoline-2-thiones 2a–c
To a solution of 4-iodoaniline, 3-iodoaniline or 2-iodoaniline (12 g, 54.8 mmol) were added 100 mL of triethylamine and 100 mL of carbon disulphide. The solution was stirred under argon for 24 h. The solution was filtered and the yellow solid was washed with diethyl ether. The dithiocarbamate salts 1a–c were used without further purification; 1a: yield: 95%, Mp: 92 °C. 1H NMR (300 MHz) δ 1.30 (t, 9H, 3J = 7.3 Hz, CH3), 3.17 (q, 6H, 3J = 7.3 Hz, CH2), 7.43 (m, 2H, Ar), 7.53 (m, 2H, Ar), 9.45 (s, 1H, NH); 13C NMR (75 MHz) δ 8.8 (CH3), 46.0 (CH2), 88.2 (Ar), 125.2 (Ar), 137.7 (Ar), 140.8 (Ar), 214.3 (CS); 1b: yield: 97%; Mp: 100 °C; 1H NMR (300 MHz) δ 1.36 (t, 9H, 3J = 7.3 Hz, CH3), 3.23 (q, 6H, 3J = 7.3 Hz, CH2), 6.99 (t, 1H, 3J = 8.1 Hz, Ar), 7.39 (d, 1H, 3J = 8.1 Hz, Ar), 7.62 (d, 1H, 3J = 8.1 Hz, Ar), 8.10 (s, 1H, Ar), 9.40 (s, 1H, NH); 13C NMR (75 MHz) δ 8.8 (CH3), 46.0 (CH2), 93.3 (Ar), 122.8 (Ar), 129.7 (Ar), 131.8(Ar), 133.3 (Ar), 142.1 (Ar), 214.7 (CS); 1c: yield: 92%; Mp: 95 °C; 1H NMR (300 MHz) δ 1.36 (t, 9H, 3J = 7.3 Hz, CH3), 3.22 (q, 6H, 3J = 7.3 Hz, CH2), 6.85 (m, 1H, Ar), 7.31 (m, 1H, Ar), 7.78 (d, 1H, 3J = 8.1 Hz, Ar), 8.08 (d, 1H, 3J = 8.1 Hz, Ar), 9.02 (s, 1H, NH); 13C NMR (75 MHz) δ 9.2 (CH3); 46.0 (CH2); 95.0 (Ar); 114.8 (Ar); 127.7 (Ar); 128.8 (Ar); 138.8 (Ar); 142.2 (Ar); 215.5 (CS). To a solution of the dithiocarbamate salt (1a–c) (20.6 g, 52.0 mmol) was added 1 equivalent of chloroacetaldehyde (0.96 mL, 52.0 mmol). The solution was stirred for 12 h at rt and 9/10 of the solvent was evaporated in vacuo. The mixture was added to 15 mL of H2SO4 at 0 °C and stirred for further 10 minutes. The solution was extracted with CH2Cl2 (3 × 50 mL), washed with water (3 × 20 mL) and dried over MgSO4. The precipitate was washed with ethanol. Brown powders were obtained.
C), 128.4 (2Ar), 132.1 (CC), 138.3 (Ar), 138.9 (2Ar), 214.5 (CS); HRMS (ESI) calcd for C9H6INNaS2 [M + Na]+: 341.88786. Found: 341.8882; anal calcd for C9H6INS2: C, 33.87; H, 1.89; N, 4.39. Found: C, 33.68; H, 1.82; N, 4.38.
C), 126.1 (CC), 130.8 (Ar), 132.1 (Ar), 135.3 (Ar), 138.3 (Ar), 139.3 (Ar), 188.7 (CS); HRMS (ESI) calcd for C9H6INNaS2 [M + Na]+: 341.88786. Found: 341.8881. Anal calcd for C9H6INS2: C, 33.87; H, 1.89; N, 4.39. Found C, 33.51; H, 1.69; N, 4.42.
C), 129.2 (CC), 129.8 (Ar), 131.4 (2Ar), 140.4 (Ar), 141.5 (Ar), 189.0 (CS); HRMS (ESI) calcd for C9H6INNaS2 [M + Na]+: 341.88786. Found 341.8877. Anal calcd for C9H6INS2: C, 33.87; H, 1.89; N, 4.39. Found: C, 33.76; H, 1.69; N, 4.44.
Synthesis of bicycles 3a–c
To a −10 °C cooled solution of thiazoline (2a–c) (1 g, 3.12 mmol) in 80 mL of dry THF under nitrogen was added a solution of LDA prepared from diisopropylamine (0.66 ml, 4.8 mmol) and 1.6 M n-BuLi (2.93 mL, 4.8 mmol) in 10 mL of dry THF. After stirring for 30 min at −10 °C, sulphur S8 (150 mg, 4.8 mmol) was added and the solution was stirred for an additional 30 min. A solution of LDA diisopropylamine (0.88 mL, 6.24 mmol) and n-BuLi (3.9 mL, 6.24 mmol) in 15 mL of dry THF was added. The mixture was stirred for 3 hours and S8 (200 mg, 6.24 mmol) was added. After 30 min, triphosgene (1.11 g, 3.7 mmol) was added to the reaction mixture. The reaction was stirred overnight and water (15 mL) was slowly added. The solvent was evaporated in vacuo. Dichloromethane (50 mL) was added and the solution was washed with water (3 × 20 mL) and dried over MgSO4. The concentrated solution was purified by chromatography on silica gel using CH2Cl2–petroleum ether as the eluent. Brown powders were obtained for 3a and 3c. 3b was not isolated, and the crude oil was used without further purification.C), 127.1 (CC), 128.3 (2Ar), 137.2 (Ar), 139.6 (2Ar), 186.1 (CS), 188.1 (CO); IR ν(CS): 1261 cm−1, ν(CO): 1695 cm−1; HRMS (ESI) calcd for C10H4INOS4 [M + H]+: 409.82933. Found: 409.8296; anal. calcd for C10H4INOS4: C, 29.34; H, 0.99; N, 3.42. Found: C, 29.68; H, 1.06; N, 3.50.
C), 112.6 (CC), 131.1 (Ar), 131.7 (Ar), 138.8 (Ar), 139.4 (Ar), 143.7 (Ar), 191.6 (CS), 192.1 (C0); IR ν(CS): 1289 nm, IR ν(CO): 1660 nm; HRMS (ESI) calcd for C10H4INOS4 [M + H]+: 431.81127. Found: 431.8110; anal. calcd for C10H4INOS4: C, 29.34; H, 0.99; N, 3.42. Found: C, 29.65; H, 0.92; N, 3.21.
Synthesis of BIP-BTTT 4a–c
A solution of thiazoline-thione 3a–c (243 mg, 0.59 mmol) (crude compound for 3b) in 30 mL of toluene was refluxed for 16 h. 80% of the solvent was removed in vacuo and the concentrated solution was filtered and the precipitate was washed with ethanol. Dark purple powders were obtained. Crystals of 4c of sufficient quality for X-ray diffraction were obtained by slow evaporation of CHCl3 solution.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 640), 358 (11500), 520 (2550); anal. calcd for [C18H8I2N2S6 + toluene (8/1)]: C, 31.93; H, 1.28; N, 3.95. Found: C, 32.08; H, 1.27; N, 3.95.
060), 359 (18370), 512 (3420); HRMS (ESI) calcd for C18H9N2IS6 [M + Na]+: 720.69935. Found 720.6993. Analysis calcd for [C18H8I2N2S6 + toluene (8/1)]: C, 31.93; H, 1.28; N, 3.95. Found: C, 32.04; H, 1.32; N, 4.18.
670), 358 (14928), 511 (2270); HRMS (ESI) calcd for C18H9N2IS6 [M]−: 697.71068. Found: 697.7107; analysis calcd for [C18H8I2N2S6 + toluene (8/1)]: C, 31.93; H, 1.28; N, 3.95. Found: C, 31.77; H, 1.25; N, 3.96.
640), 358 (11500), 520 (2550); anal. calcd for [C18H8I2N2S6 + toluene (8/1)]: C, 31.93; H, 1.28; N, 3.95. Found: C, 32.08; H, 1.27; N, 3.95.
060), 359 (18370), 512 (3420); HRMS (ESI) calcd for C18H9N2IS6 [M + Na]+: 720.69935. Found 720.6993. Analysis calcd for [C18H8I2N2S6 + toluene (8/1)]: C, 31.93; H, 1.28; N, 3.95. Found: C, 32.04; H, 1.32; N, 4.18.
670), 358 (14928), 511 (2270); HRMS (ESI) calcd for C18H9N2IS6 [M]−: 697.71068. Found: 697.7107; analysis calcd for [C18H8I2N2S6 + toluene (8/1)]: C, 31.93; H, 1.28; N, 3.95. Found: C, 31.77; H, 1.25; N, 3.96.
Crystallography
Data were collected on a D8 VENTURE Bruker AXS diffractometer with graphite-monochromated Mo-Kα radiation (λ = 0.71073 Å) for 2b, 3a, 3c and 4c. The structures were solved by a dual-space algorithm using the SHELXT program,17 and then refined with full-matrix least-squares methods based on F2 (SHELXL).18 All non-hydrogen atoms were refined with anisotropic atomic displacement parameters. H atoms were finally included in their calculated positions. Concerning 3a, the use of the Platon/TwinRotMat19 routine allowed the presence of a twinning in the measured crystal to be detected. Satisfactory final structural refinement has been performed considering the presence of such a twinning and in the hklf5 file format, leading to a refined twin ratio of 0.19. Crystallographic data on X-ray data collection and structure refinements are given in Table 3.| Compound | 2b | 3a | 3c | 4c |
|---|---|---|---|---|
| Formula | C9H6INS2 | C10H4INOS4 | C10H4INOS4 | C18H8I2N2S6 |
| FW (g mol−1) | 319.17 | 409.28 | 409.28 | 698.42 |
| Crystal system | Monoclinic | Monoclinic | Monoclinic | Triclinic |
| Space group | C2/c | P21/n | P21/c |
P |
| a (Å) | 8.7777(10) | 4.1078(7) | 8.1394(7) | 12.5260(18) |
| b (Å) | 11.1568(15) | 24.675(5) | 6.9798(7) | 14.352(2) |
| c (Å) | 21.241(3) | 13.943(3) | 22.487(2) | 14.522(2) |
| α (°) | 90 | 90 | 90 | 117.141(4) |
| β (°) | 96.632(4) | 118.785(7) | 94.788(3) | 95.478(5) |
| γ (°) | 90 | 90 | 90 | 100.638(5) |
| V (Å3) | 2066.3(5) | 1238.6(4) | 1273.1(2) | 2234.7(6) |
| T (K) | 150(2) | 150(2) | 150(2) | 150(2) |
| Z | 8 | 4 | 4 | 4 |
| D calc (g cm−3) | 2.052 | 2.195 | 2.135 | 2.076 |
| μ (mm−1) | 3.454 | 3.238 | 3.151 | 3.384 |
| Total refls. | 8223 | 11687 |
14751 |
51086 |
| Abs. corr. | Multi-scan | Multi-scan | Multi-scan | Multi-scan |
| Uniq. refls. (Rint) | 2344 (0.0274) | — | 2904 (0.0404) | 10225 (0.1050) |
| Uniq. refls. (I > 2σ(I)) | 2188 | 8472 | 2675 | 6098 |
| R 1, wR2 | 0.0307, 0.0974 | 0.0758, 0.1622 | 0.0306, 0.0814 | 0.1160, 0.3164 |
| R 1, wR2 (all data) | 0.0336, 0.1095 | 0.1094, 0.1758 | 0.0337, 0.0834 | 0.1793, 0.3626 |
| GoF | 1.101 | 1.042 | 1.054 | 1.079 |
Theoretical modeling
Electrostatic surface potential calculations were carried out on the optimized geometry of the molecules (with density functional theory using the Gaussian 09 revision D.01 software, the B3LYP functional and the 6-31+G** basis set for all the atoms and the LANLdp basis set for iodine). GaussView 5.0.9 was used to generate the figures.Conflicts of interest
There are no conflicts to declare.Notes and references
- S. Sutton, C. Risko and J. L. Brédas, Chem. Mater., 2016, 28, 3–16 CrossRef.
- G. R. Desiraju, J. Am. Chem. Soc., 2013, 135, 9952–9967 CrossRef CAS PubMed.
- R. Gleiter, G. Haberhauer, D. B. Werz, F. Rominger and C. Bleiholder, Chem. Rev., 2018, 118, 2010–2041 CrossRef CAS PubMed.
- G. R. Desiraju, Cryst. Growth Des., 2011, 11, 896–898 CrossRef CAS.
- E. D. Głowacki, M. Irimia-Vladu, S. Bauer and N. S. Sariciftci, J. Mater. Chem. B, 2013, 1, 3742–3753 RSC.
- K. S. Kim, P. Tarakeshwar and J. Y. Lee, Chem. Rev., 2000, 100, 4145–4185 CrossRef CAS PubMed.
- (a) G. Cavallo, P. Metrangolo, R. Milani, T. Pilati, A. Priimagi, G. Resnati and G. Terraneo, Chem. Rev., 2016, 116, 2478–2601 CrossRef CAS PubMed; (b) L. C. Gilday, S. W. Robinson, T. A. Barendt, M. J. Langton, B. R. Mullaney and P. D. Beer, Chem. Rev., 2015, 115, 7118–7195 CrossRef CAS PubMed.
- A. S. Mahadevi and G. N. Sastry, Chem. Rev., 2016, 116, 2775–2825 CrossRef CAS PubMed.
- (a) Y. Le Gal, N. Bellec, F. Barrière, R. Clérac, M. Fourmigué, V. Dorcet, T. Roisnel and D. Lorcy, Dalton Trans., 2013, 42, 16672–16679 RSC; (b) Y. Le Gal, D. Ameline, N. Bellec, A. Vacher, T. Roisnel, V. Dorcet, O. Jeannin and D. Lorcy, Org. Biomol. Chem., 2015, 13, 8479–8486 RSC; (c) Y. Le Gal, M. Rajkumar, A. Vacher, V. Dorcet, T. Roisnel, M. Fourmigué, F. Barrière, T. Guizouarn and D. Lorcy, CrystEngComm, 2016, 18, 3925–3933 RSC.
- A. Filatre-Furcate, T. Higashino, D. Lorcy and T. Mori, J. Mater. Chem. C, 2015, 3, 3569–3573 RSC.
- (a) K. Iijima, Y. Le Gal, T. Higashino, D. Lorcy and T. Mori, J. Mater. Chem. C, 2017, 5, 9121–9127 RSC; (b) K. Iijima, Y. Le Gal, D. Lorcy and T. Mori, RSC Adv., 2018, 8, 18400–18405 RSC.
- C. Wang, H. Dong, W. Hu, Y. Liu and D. Zhu, Chem. Rev., 2012, 112, 2208–2267 CrossRef CAS PubMed.
- Y. Le Gal, D. Lorcy, O. Jeannin, F. Barrière, V. Dorcet, J. Lieffrig and M. Fourmigué, CrystEngComm, 2016, 18, 5474–5481 RSC.
- M. L. Kaplan, R. C. Haddon, F. B. Bramwell, F. Wudl, J. H. Marshall, D. O. Cowan and S. Gronowitz, J. Phys. Chem., 1980, 84, 427–431 CrossRef CAS.
- M. Arca, M. C. Aragoni, F. A. Devillanova, A. Garau, F. Isaia, V. Lippolis, A. Mancini and G. Verani, Bioinorg. Chem. Appl., 2006, 58937 Search PubMed.
- P. Auffinger, F. A. Hays, E. Westhof and P. S. Ho, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 16789–16794 CrossRef CAS PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- A. L. Spek, Acta Crystallogr., Sect. A: Found. Crystallogr., 1990, 46, C34 Search PubMed.
Footnote |
| † CCDC 1872721–1872724. For crystallographic data in CIF or other electronic format see DOI: 10.1039/c8ce02046a |
| This journal is © The Royal Society of Chemistry 2019 |