
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Conundrum of γ glycine nucleation revisited: to stir or not to stir?†‡
Maria J.
Vesga
 a,
David
McKechnie
ab,
Paul A.
Mulheran
a,
Karen
Johnston
a and
Jan
Sefcik
*ac
a,
David
McKechnie
ab,
Paul A.
Mulheran
a,
Karen
Johnston
a and
Jan
Sefcik
*ac
aDepartment of Chemical and Process Engineering, University of Strathclyde, 75 Montrose Street, Glasgow, G1 1XJ, UK. E-mail: jan.sefcik@strath.ac.uk
bDoctoral Training Centre in Continuous Manufacturing and Advanced Crystallisation, University of Strathclyde, Glasgow, G1 1RD, UK
cEPSRC Future Manufacturing Hub in Continuous Manufacturing and Advanced Crystallisation, University of Strathclyde, Glasgow, G1 1RD, UK
First published on 15th March 2019
Abstract
Glycine polymorphism presents a conundrum: while the metastable α form of glycine typically crystallises in bulk cooling crystallisation from aqueous solution, both the highly unstable β and stable γ forms can be selectively crystallised in small scale cooling or evaporative experiments, without any additives, cosolvents or external fields. Small scale experiments in microwells or droplets differ from bulk crystallisation in some key aspects: absence of agitation, presence of large (and often very particular) surface areas per crystallisation volume, and ability to reach very high supersaturations. In this work we investigated effects of agitation on polymorphic outcomes in glycine crystallisation from aqueous solutions across a wide range of supersaturations at mL scale under quiescent conditions with and without a PTFE-coated magnetic stirrer (without any stirring) as well as under stirred conditions (with agitation supplied by the stirrer). In the absence of stirring, γ was predominant at higher glycine concentrations, which indicates that γ is more likely to nucleate than α in highly supersaturated aqueous solutions under quiescent conditions. Intriguingly, we found that under stirred conditions α was predominant at all concentrations and temperatures investigated. The effect of stirring on the preference for α glycine polymorphism cannot be fully explained by secondary nucleation alone. Instead, primary nucleation of glycine (at least of metastable forms) is strongly enhanced by stirring, in agreement with previous observations of shear effect on primary nucleation of glycine, and it is likely that similar effects play a role in other polymorphic systems of pharmaceutical interest.
1. Introduction
Formation of different polymorphs in pharmaceutical crystallisation is of great interest since the solid form affects the downstream processability of drug substance and performance of the final drug product, such as bioavailability, dissolution and stability.1–3 Many approaches have been proposed to control polymorphism, including solvents, concentration, temperature, control of supersaturation profiles, additives and interfaces.1,2,4–6 Initial crystallisation screening and process development is often performed at small scale where mixing and agitation is ill-defined or even absent and therefore these effects are often underestimated or overlooked. However, there are numerous examples where agitation plays profound role in determining polymorphic outcome in crystallisation of pharmaceuticals and other molecular systems, such as carbamazepine, stearic acid, L-glutamic acid, m-hydroxybenzoic acid7 or glycine.8–11 In industrial applications it is vital to understand the impact of process conditions such as agitation on polymorphism in order to scale up crystallisation processes properly accounting for differences in agitation between lab-based, pilot scale and plant scale processes.Glycine is used to treat a variety of health issues, including anxiety, schizophrenia and insomnia.12 In aqueous solution it is present as a zwitterionic species (+NH3CH2COOH−) and forms mesoscale clusters13 and crystallises in several different polymorphic forms.14–16 There are three glycine polymorphs that could form under ambient conditions: α, β and γ. The γ form is the most stable polymorph at ambient conditions, followed by the metastable forms α and then β.17–19 There have been numerous papers reporting glycine solubility, and while there is some uncertainty in the previous literature regarding α solubility,20 there is a major disagreement among reported solubility data for γ11,21,22 (see Fig. 1). The least stable polymorph β, has been observed to be rapidly replaced (within a few minutes) by α in agitated aqueous solutions.11,23 α is a metastable polymorph and under agitation in aqueous solutions it is very slowly replaced by the stable γ polymorph (reported to start after 35 hours24). Mechanisms of these replacements are not fully understood: they may include primary nucleation (more stable polymorph nucleates in supersaturated solution suspending less stable polymorph), secondary nucleation (crystals of one polymorph inducing nucleation of another polymorph), competitive nucleation and growth (e.g., two polymorphs nucleate but the less stable one grows faster that the more stable one), or solid–solid transformation. For glycine, relative nucleation rates of different polymorphs are not known, but it has been reported that γ can induce secondary nucleation of either α or γ.25 Relative growth rates of β vs. other polymorphs are not known, but it has been shown that α grows faster than γ in aqueous solutions.26 It was also shown that β can undergo solid–solid transformation to α or γ forms.27
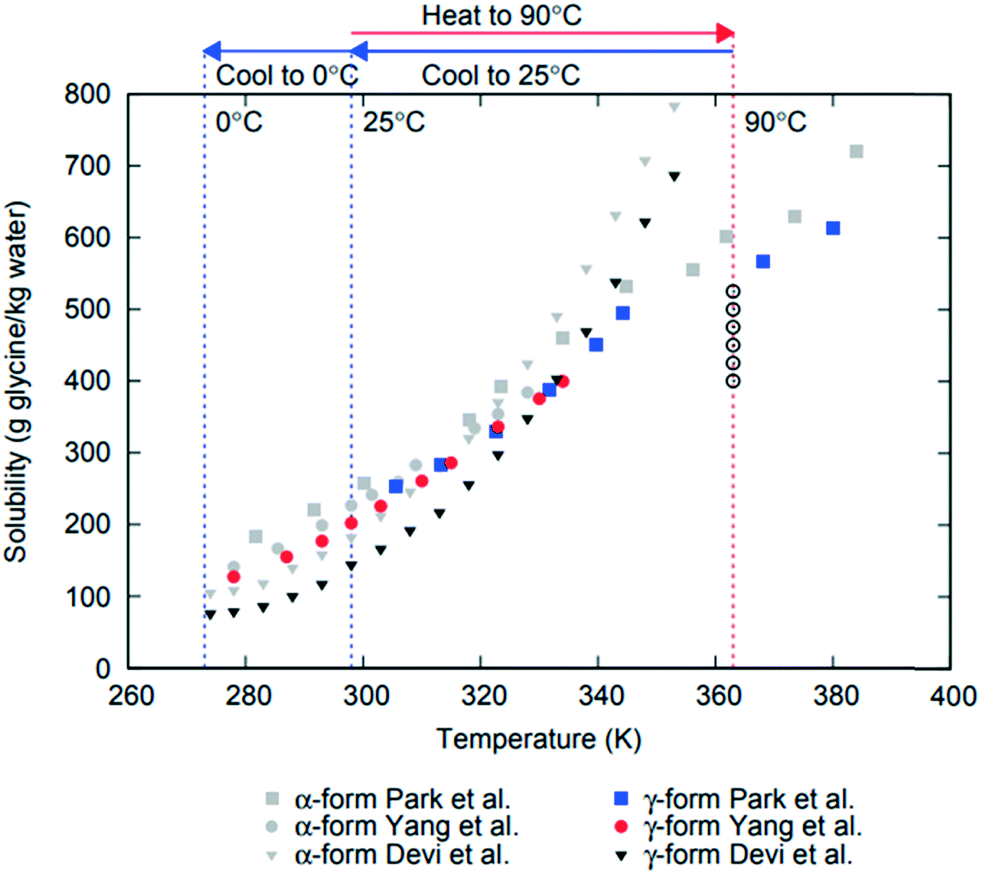 | ||
| Fig. 1 Reported solubility data for α and γ polymorphs of glycine from Devi et al.,11 Yang et al.21 and Park et al.22 Glycine solution compositions used in this work are shown as open circles at 363 K (90 °C) where undersaturated solutions were prepared. | ||
Many strategies have been previously applied to achieve selective crystallisation of glycine polymorphs using pH change, additives or cosolvents, which can accelerate or inhibit either nucleation or growth in order to steer the crystallisation outcome towards a desirable form.24,28,29 Even though it has been reported that α is invariably obtained in conventional cooling crystallisation from pure water under agitated conditions,17 all three polymorphs can be crystallised from purely aqueous solutions under suitable conditions.11,16,30 For example, γ was obtained via slow evaporation from microdroplets31 and it was shown that γ was more likely to crystallise than α at higher supersaturations. A study of glycine crystallisation under quiescent conditions at a lower concentration resulted in α in the bulk,32 although a mixture of α and γ crystals was observed upon evaporation of thin films on beaker walls. Surovtsev et al. performed a detailed study of the formation of glycine crystalline and amorphous solid phases using either controlled or quench cooling of glycine aqueous solutions to subzero temperatures.33
Recent works revealed that agitation, such as shear flow or stirring, can affect the primary nucleation and steer selective polymorph crystallisation. Studies using Couette cells and capillary flow devices reported a strong effect of shear on the rate of primary nucleation of α glycine.34 In a study by Devi et al., glycine crystallisation was performed under agitation at 0 °C and α crystallised at higher concentrations, while pure β and mixtures of β and α were obtained at lower concentrations and lower stirring rates.11 Igarashi et al. found that in agitated cooling crystallisation of glycine, γ was obtained by using a wall wetter (slurry sprinkler) setup, while α was obtained otherwise.35 The effect of stirring as a controlling factor has been studied in competitive crystallisation of polymorphic forms in different systems. Cashell et al.9 studied the crystallisation in supersaturated solutions of L-glutamic acid and found that metastable α is obtained using either continuous or pulsed agitation during slow cooling, whereas the stable β polymorph was preferentially obtained for both fast cooling with agitation and for slow cooling without stirring. Tahri10 reported that supersaturated solutions of L-glutamic acid under stirring conditions show the preferential formation of the metastable α form while the stable β form was obtained under stagnant conditions. The effect of stirring on m-hydroxybenzoic acid solutions found that the proportion of the stable form decreases under intermediate agitation rates, accompanied by a large reduction in nucleation time of the metastable form.7 The effect of stirring conditions on cooling crystallisation of carbamazepine from anhydrous ethanol was investigated by Sypek et al.8 In this study, crystals of form II were formed and then slowly transformed to crystals of form III under quiescent conditions. In the case of sufficiently vigorous stirring, the induction times observed were clearly defined by the onset of turbidity which was due to formation of a large number of small form III crystals.
It is clear that stirring can promote or hinder the formation of the stable polymorph. In the present work, we investigate the effects of stirring on the polymorphic crystallisation of glycine from aqueous solutions. To elucidate these effects, isothermal polymorph formation for either quiescent or stirring conditions was observed over a range of glycine concentrations and two different crystallisation temperatures. Understanding effects of agitation on polymorph control will aid rational design and scale up of industrial crystallisation processes.
2. Experimental methods
2.1. Preparation of glycine stock solutions
Glycine solutions with a range of concentrations of 400–525 g glycine per kg water (referred to hereafter as g per kg water) were prepared at a high temperature (90 °C) where they were all undersaturated and then cooled to a desired crystallisation temperature (either 0 °C or 25 °C). Solid commercial glycine powder (≥99% electrophoresis from Sigma) and deionized water (Milli-Q, 18.2 MΩ cm) were used. Required amounts of glycine and water were weighted at room temperature, transferred into a 100 mL glass bottle and sealed. The solution was heated and stirred at 600 rpm for 1 hour at 90 °C ± 1 °C using a hotplate magnetic stirrer (IKA RCT Basic) supplied with an external temperature probe. Aliquots of 0.65 mL of stock solution were then transferred into preheated vials (VWR; screw vial, ID = 1156587 411.6 mm × 32 mm height, neck diameter 8 mm, total volume 1.5 mL) at 90 °C and sealed with parafilm and lids. Sealed vials were kept under thermostatted conditions for 15 min at 90 °C using a Polar Bear Plus Crystal. The Polar Bear is a precision heating and cooling platform produced by Cambridge Reactor Design that uses interchangeable plate attachments to allow for accurate (±0.1 °C) temperature control for a range of vessels from vials to round bottom flasks. Supersaturated solutions were then obtained by cooling the vials from 90 °C to either 0 °C or 25 °C, using a controlled cooling rate in Polar Bear. Once solutions reached the final temperature, they were kept under either quiescent or stirred conditions (see below). The temperature of preparation of the glycine solutions was chosen by taking into account the reported solubility for α and γ glycine11,21,22 in order to make sure that all solutions were undersaturated with respect to the most stable polymorph at the solution preparation temperature. We note that there are very large discrepancies in previously reported data on γ solubility as can be seen in Fig. 1, which summarises the solubility for α and γ forms of glycine. It also shows the concentration and temperature ranges used in this study based on a conservative estimate of likely temperature dependence of γ glycine solubility.2.2. Crystallisation under quiescent conditions
Crystallisation experiments under quiescent conditions (without stirring) were performed using controlled cooling of glycine solutions at all concentrations investigated. The following protocol describes the crystallisation of the solutions when controlled cooling to two different temperatures was used. Controlled cooling (Fig. 2, setup 1a and b) of 28 vials was performed for each concentration over three different periods. During period I, solutions were cooled from 90 °C to either 0 °C or 25 °C using Polar Bear with a cooling rate of −1.5 °C min−1. During period II, the samples were kept isothermal in Polar Bear at either 0 °C or 25 °C for 3 hours to monitor crystal formation. For period III (after the 3 hours), the vials at 25 °C were transferred to an incubator (Stuart Scientific Incubator S160D) at 25 ± 0.5 °C. The vials at 0 °C were heated to 25 °C at a rate of 1.5 °C min−1 and then transferred to the incubator at 25 °C. In both cases, the vials were kept in the incubator for two weeks to monitor the crystallisation. | ||
| Fig. 2 Diagrams illustrate the different temperature profiles used in this study. Setup 1 is quiescent conditions and setup 2 is stirring conditions, each using controlled cooling to (a) 0 °C, and (b) 25 °C. | ||
Crystallisation was monitored visually throughout all experiments. During periods I and II for all setups, the crystals were isolated at the end of the period to avoid disturbing the other vials and potentially inducing nucleation. During the longer period III of setup 1 (2 weeks) the vials were checked for crystallisation daily (except at weekends) and crystals were isolated once they were observed.
2.3. Crystallisation under stirring conditions
A protocol including controlled cooling and stirring of the solutions at the crystallisation temperature is shown in Fig. 2 setup 2a and b. First, for each concentration, 15 vials with a stirring bar inside (VWR micro magnetic bars, PTFE covered, round, smooth surface, length 7 mm and diameter 2 mm), were cooled from 90 °C at a rate of −1.5 °C min−1 to either 0 °C or 25 °C using Polar Bear (period I). When the final temperature (0 °C or 25 °C) was reached, all the vials were swiftly transferred to a stirring plate (2mag, Magnetic stirrer MIXdrive with control unit MIXcontrol, 15 stirring points) placed either immersed in an ice bath at 0 ± 1 °C or inside an incubator at 25 ± 0.5 °C. Two different stirring speeds, 1600 rpm and 100 rpm, and a control at 0 rpm, were used (period II). After transfer, crystallisation was monitored for 3 hours (period II). After 3 hours, the vials at 25 °C were kept at the same temperature and the vials at 0 °C were swiftly transferred to an incubator at 25 ± 0.5 °C and left for 24 hours (period III). Crystallisation was monitored visually throughout all experiments as described above. In setup 2, period III was 24 hours long and crystals were isolated at the end of the period.2.4. Characterization of glycine crystals
Immediately after finishing each crystallisation stage (end of periods I and II in setup 1 and I, II and III in setup 2), all crystals were removed from the vials and placed onto filter paper to remove excess liquid. For period III in setup 1 crystals were removed as they were detected (checked daily, except weekends). The crystals were dried for one day at room temperature and then ground to a fine powder before analysis. Regarding possible transformations during drying, our crystals are dried under stagnant conditions and it is unlikely that a polymorphic transformation from alpha to gamma would occur. In the study by Yang et al.,24 the glycine crystals were in an agitated slurry, which only started to transform after 35 hours. Since our crystals were left to dry for a day under stagnant conditions, it is unlikely that any transformation would occur.The analysis of powder samples was carried out using both Fourier transform infrared spectroscopy (FTIR) and X-ray powder diffraction (XRPD). FTIR spectra were obtained with an ABB MB3000 spectrometer. Absorbance spectra were obtained and averaged over 32 scans at a resolution of 8 cm−1 in the range of 500 to 4000 cm−1 with a detector gain of 80.68. XRPD patterns were obtained by placing 10–50 mg of sample on a 28-well plate supported on a polyimide (Kapton 7.5 μm thickness) film. Data were collected on a Bruker AXS D8-Advance II transmission diffractometer equipped with θ/θ geometry, primary monochromated radiation (Cu Kα1, λ = 1.54056 Å), a Vantec 1D position sensitive detector (PSD) and an automated multi-position x–y sample stage. Data were collected in the range 4–35° 2θ with a 0.015° 2θ step size and 1 s step-1 count time. All FTIR spectra and XRPD patterns were obtained at ambient temperature. The commercial glycine powder was also analysed and it was found to be composed of the α form (Fig. 4a and b), although a trace amount of γ form was also detected by XRPD. With this in mind, and taking into account glycine solubilities (Fig. 1), we ensured the complete dissolution of glycine at 90 °C at all solution concentrations investigated to avoid potential seeding with γ glycine. Crystals were viewed using a Brunel BMDZ series optical microscope. Images were captured from the optical microscope using a Canon EOS 1200D digital SLR camera equipped with a EF-S 18-55 mm IS II lens.
3. Results and discussion
3.1. Polymorphic identification
The different glycine solid forms and corresponding morphologies obtained in this study are shown in Fig. 3. Needle-like γ crystals were most often crystallised under quiescent conditions over the range of conditions investigated. Fig. 3a shows a typical outcome of numerous γ crystals obtained from a 500 g per kg water solution at 0 °C (during period II in setup 1a). The needle-like γ glycine crystals observed here are similar to those previously reported32,36 although other morphologies, such as trigonal pyramidal were also reported.30 When α crystals formed under quiescent conditions they had a large prismatic morphology. Fig. 3b shows a single α crystal obtained from a 475 g per kg water solution at 25 °C (during period III in setup 1a). In some quiescent samples a mixture of both prismatic α crystals and needle-like γ crystals was obtained. Fig. 3c shows a single α crystal together with a mass of numerous γ crystals obtained from a 500 g per kg water solution at 25 °C (during period II in setup 1b). However, under stirring conditions, numerous α crystals formed rapidly when stirring started after the crystallisation temperature was reached. Fig. 3d shows a mass of precipitated α crystals obtained from a 525 g per kg water solution at 0 °C that turned milky white after few seconds of stirring at 1600 rpm (during period II in setup 2a). | ||
| Fig. 3 Optical microscopy images of different crystal morphologies produced under different crystallisation conditions. a) Needle-like γ crystals from quiescent conditions, period II, setup 1a, 500 g per kg water, 0 °C; b) single α crystal from quiescent conditions, period III, setup 1a, 475 g per kg water, 25 °C; c) single α crystal and numerous γ crystals from quiescent conditions, period II, setup 1b, 500 g per kg water, 25 °C; d) precipitated α crystals from stirring conditions, period II, setup 2a, 525 g per kg water, 0 °C, 1600 rpm. | ||
In order to unambiguously identify the polymorphic form present in the solids obtained under each crystallisation condition, FTIR and XRPD were used. Fig. 4 shows typical FTIR spectra (a) and XRPD patterns (b) obtained for each morphology shown in Fig. 3. The characteristic IR peaks at around 910 cm−1 for α, at around 930 cm−1 for γ and a common peak at 887 cm−1 for both forms (Fig. 4a) were selected for solid form identification.24 The commercial glycine powder used to prepare the solutions shows peaks corresponding to α. Characteristic XRPD peaks at 2θ of 19.5° and 29.7° correspond to α crystals, while those of 21° and 25.3° correspond to γ crystals (cf. International Center for Diffraction Data (ICDD) files 00-032-1702 and 02-088-4306 for α and γ, respectively). The XRPD pattern obtained for samples containing mixtures of needle-like prismatic crystals shows the presence of characteristic peaks of both polymorphs α and γ (Fig. 4b). The XRPD pattern obtained for the commercial glycine powder showed characteristic peaks of α and very small γ peaks, indicating a minor γ impurity. The β form can be identified by an XRPD peak at 2θ of 18°,11 but this peak is not observed in any of our samples.
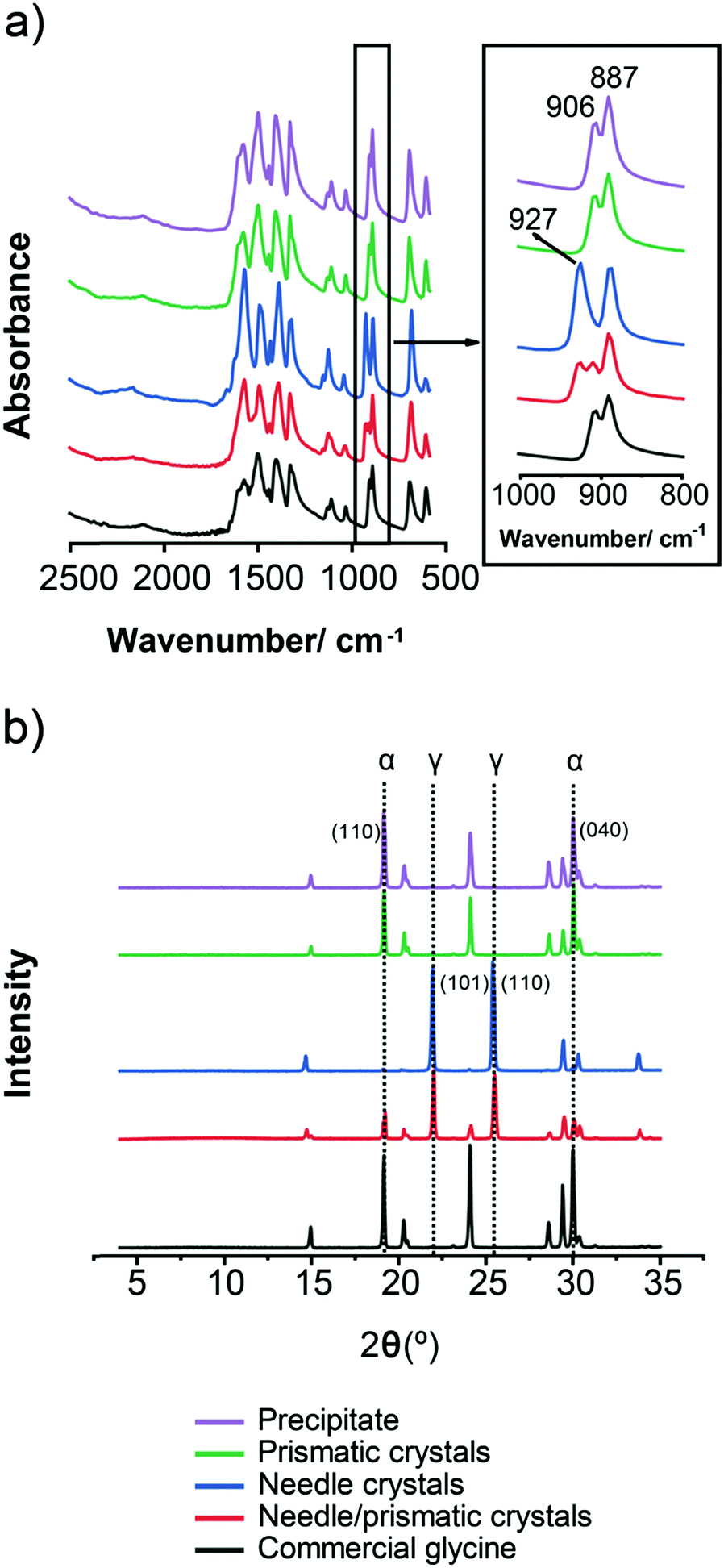 | ||
| Fig. 4 (a) FTIR spectra (b) XRPD spectra of glycine solids. FTIR and XRPD both show that precipitate, prismatic crystals and commercial glycine are mainly α, whereas needles are γ, and needles/prismatic crystals are a mixture of α and γ. | ||
3.2. Glycine crystallisation under quiescent conditions
The effect of the crystallisation temperature and concentration on glycine polymorphic crystallisation was first studied under quiescent conditions.Crystallisation was monitored across the three different periods I, II and III (see setup 1 in Fig. 2). No crystallisation was observed during the cooling (period I) before the final crystallisation temperature (either 25 °C or 0 °C) was reached. During the following isothermal period II, up to about 50% of vials crystallised at 0 °C, while up to 30% of vials crystallised at 25 °C at the highest concentrations of 525 g per kg water. There was some further crystallisation observed during period III, but even after 2 weeks at 25 °C many vials still had no crystals. For both crystallisation temperatures, the fraction of vials crystallised decreased drastically at lower concentrations, as shown in Fig. 5a and b. This is expected as the supersaturation is higher at higher concentrations and lower temperatures.
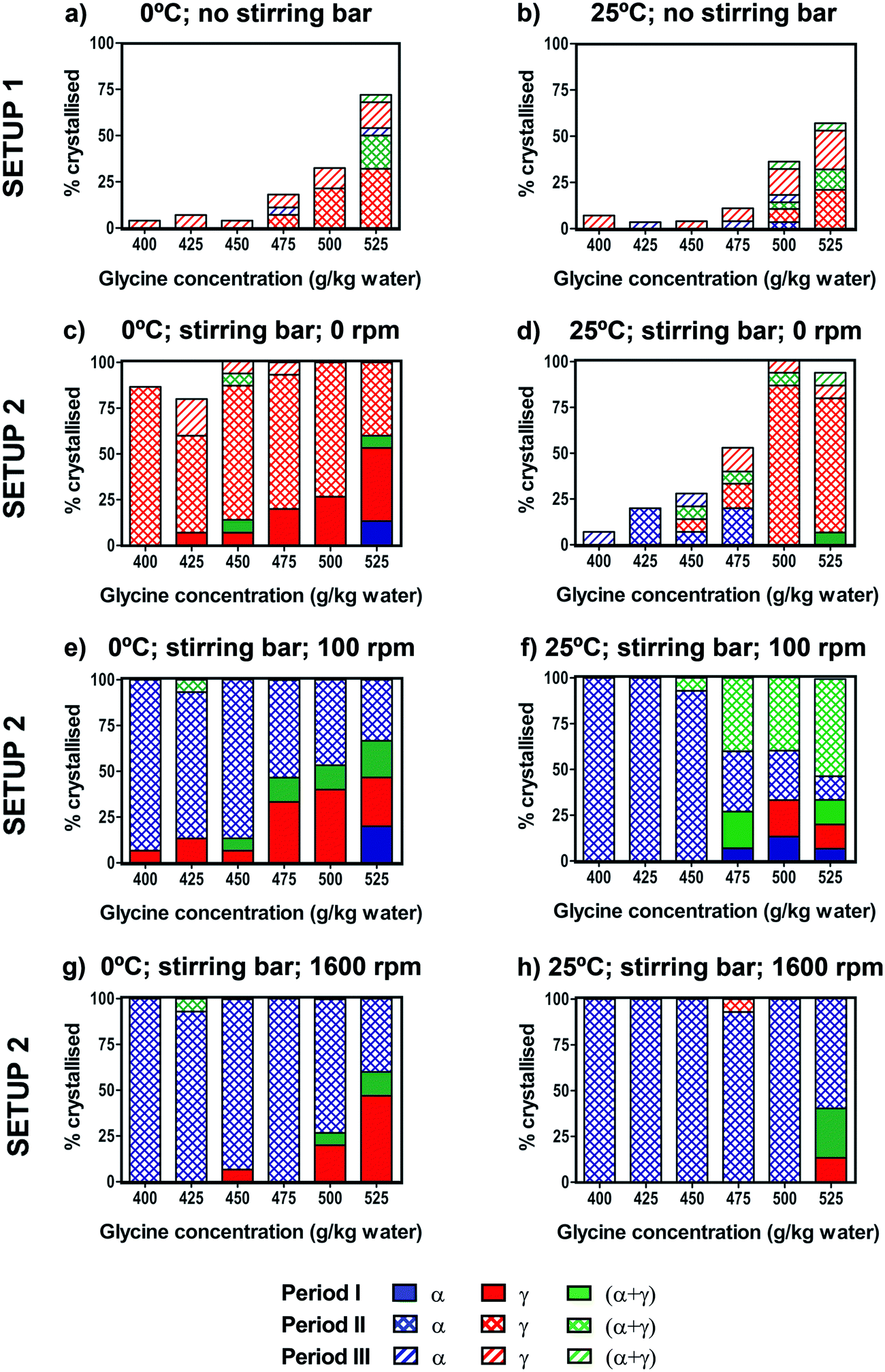 | ||
| Fig. 5 Crystallisation of glycine solutions under quiescent conditions (setup 1) cooled to 0 °C (a) and 25 °C (b). For each concentration in setup 1, the percentage was calculated over 28 vials. Crystallisation of glycine solutions under stirring conditions (setup 2) cooled to 0 °C (c, e and g) or 25 °C (d, f and h). The stirring rates were 0 rpm (with stirring bar present) (c and d), 100 rpm (e and f) and 1600 rpm (g and h). For each concentration in setup 2, the percentage was calculated over 15 vials. | ||
For both crystallisation temperatures and all concentrations, the γ form was predominantly obtained, with some vials forming α + γ mixtures and a small number of vials containing the pure α form. Results of our glycine crystallisation experiments under quiescent conditions show that formation of γ glycine is favoured at the range of concentrations and temperatures investigated. It was previously observed that α glycine formation was preferred at lower concentrations,37,38 where glycine solutions with concentrations up to 400 g per kg water were prepared at 60 °C. We note that in this work glycine solutions were prepared at 90 °C in order to access higher supersaturations, and that thermal history of solutions could play a role in both nucleation kinetics and polymorphic outcomes.39,40 Our results, showing a preference for γ over α at higher supersaturations, are in agreement with observations from evaporating droplets.31 This shows that both α and γ nucleate under quiescent conditions at concentrations and temperatures investigated. The transformation of α to γ is expected to be very slow in the absence of agitation compared to the maximum time our crystals spent in solution, and α grows faster than γ,26,36 thus depleting supersaturation and suppressing further nucleation and growth of γ. Therefore, it can be concluded that γ nucleation is likely to be faster than that of α. While we have never observed any β, it cannot be ruled out that some β nucleates and rapidly transforms to either α or γ. It is also unlikely that a polymorphic transformation from α to γ occurred during the drying period as the residual solution evaporated in less than one day.
3.3. Glycine crystallisation under stirring conditions
The effect of stirring on glycine crystallisation was studied using controlled cooling over the range of concentrations. Fig. 5c–h show the percentage of vials crystallised for solutions cooled to 0 °C or 25 °C and stirred at either 0 rpm (control with stirring bar present), 100 rpm or 1600 rpm. In order to control for a possible effect of the stirring bar on the crystallisation process, experiments were performed with the stirring bar present but not stirring (0 rpm). We found that for both 0 °C and 25 °C the presence of a stirring bar (Fig. 5c and d) results in a significant increase in the number of samples crystallised compared to samples without a stirring bar (Fig. 5a and b). In Table 1 we see that in experiments with a stirring bar 94% and 50% crystallised for 0 °C and 25 °C respectively, whereas without a stirring bar only 23% and 11% crystallised for 0 °C and 25 °C, respectively. However, the presence of the stirring bar does not significantly change the polymorphic outcomes compared to the experiments without the stirring bar, as in both cases γ predominantly forms. It is likely that presence of PTFE coated stirrer bar induced heterogeneous nucleation at solution-PTFE interface as it was observed that crystals tended to grow on the stirrer bar in the absence of agitation.| a) Setup 1, 0 °C | |||||
|---|---|---|---|---|---|
| Period | α | γ | α + γ | Crystallised | % crystallised |
| I | 0 | 0 | 0 | 0 | 0.0 |
| II | 0 | 17 | 5 | 22 | 13.1 |
| III | 2 | 13 | 1 | 16 | 9.5 |
| Total | 2 | 30 | 6 | 38 | 22.6 |
| Final (%) | 1.2 | 17.9 | 3.6 | 22.6 | |
| b) Setup 1, 25 °C | |||||
|---|---|---|---|---|---|
| Period | α | γ | α + γ | Crystallised | % crystallised |
| I | 0 | 0 | 0 | 0 | 0.0 |
| II | 1 | 8 | 4 | 13 | 7.7 |
| III | 3 | 1 | 2 | 6 | 3.6 |
| Total | 4 | 9 | 6 | 19 | 11.3 |
| Final (%) | 2.4 | 5.4 | 3.6 | 11.3 | |
| c) Setup 2, 0 °C, 0 rpm | |||||
|---|---|---|---|---|---|
| Period | α | γ | α + γ | Crystallised | % crystallised |
| I | 2 | 15 | 2 | 19 | 21.1 |
| II | 0 | 60 | 1 | 61 | 67.8 |
| III | 0 | 5 | 0 | 5 | 5.6 |
| Total | 2 | 80 | 3 | 85 | 94.4 |
| Final (%) | 2.2 | 88.9 | 3.3 | 94.4 | |
| d) Setup 2, 25 °C, 0 rpm | |||||
|---|---|---|---|---|---|
| Period | α | γ | α + γ | Crystallised | % crystallised |
| I | 0 | 0 | 1 | 1 | 1.1 |
| II | 7 | 27 | 3 | 37 | 41.1 |
| III | 2 | 4 | 1 | 7 | 7.8 |
| Total | 9 | 31 | 5 | 45 | 50.0 |
| Final (%) | 10.0 | 34.4 | 5.6 | 50.0 | |
| e) Setup 2, 0 °C, 100 rpm | |||||
|---|---|---|---|---|---|
| Period | α | γ | α + γ | Crystallised | % crystallised |
| I | 3 | 19 | 8 | 30 | 33.3 |
| II | 59 | 0 | 1 | 60 | 66.7 |
| III | 0 | 0 | 0 | 0 | 0.0 |
| Total | 62 | 19 | 9 | 90 | 100.0 |
| Final (%) | 68.9 | 21.1 | 10.0 | 100.0 | |
| f) Setup 2, 25 °C, 100 rpm | |||||
|---|---|---|---|---|---|
| Period | α | γ | α + γ | Crystallised | % crystallised |
| I | 4 | 5 | 5 | 14 | 15.6 |
| II | 55 | 0 | 21 | 76 | 84.4 |
| III | 0 | 0 | 0 | 0 | 0.0 |
| Total | 59 | 5 | 26 | 90 | 100.0 |
| Final (%) | 65.6 | 5.6 | 28.9 | 100.0 | |
| g) Setup 2, 0 °C, 1600 rpm | |||||
|---|---|---|---|---|---|
| Period | α | γ | α + γ | Crystallised | % crystallised |
| I | 0 | 11 | 3 | 14 | 15.6 |
| II | 75 | 0 | 1 | 76 | 84.4 |
| III | 0 | 0 | 0 | 0 | 0.0 |
| Total | 75 | 11 | 4 | 90 | 100.0 |
| Final (%) | 83.3 | 12.2 | 4.4 | 100.0 | |
| h) Setup 2, 25 °C, 1600 rpm | |||||
|---|---|---|---|---|---|
| Period | α | γ | α + γ | Crystallised | % crystallised |
| I | 0 | 2 | 4 | 6 | 6.7 |
| II | 83 | 1 | 0 | 84 | 93.3 |
| III | 0 | 0 | 0 | 0 | 0.0 |
| Total | 83 | 3 | 4 | 90 | 100.0 |
| Final (%) | 92.2 | 3.3 | 4.4 | 100.0 | |
We note that movement of vials with stirrer bar can induce nucleation and care was taken to minimise the movement of vials. Nevertheless, this effect was observed in a few cases at high concentrations when transferring the vials from Polar Bear at the end of period I to the stirring plate at the start of period II. This occurred only when a stirring bar was present, and the crystal formation happened almost instantaneously. In Fig. 5, these movement-induced cases were counted within the cooling period I. We have singled out the movement induced cases and shown them in the table in ESI.‡ On its own, fluid shear induced by stirrer movements during vial transfers would be expected to enhance nucleation of α, based on previous fluid shear experiments.41 However, as can be seen in the table shown in ESI,‡ either γ or mixtures of α and γ were observed when rapid crystallisation occurred due to vial movements, indicating that γ was present in all cases, consistent with heterogeneous nucleation.
In order to investigate the effect of stirring, stirring was switched on at the start of period II after cooling was completed and the desired crystallisation temperature was reached. 100% of vials crystallised within period II, whereas for unstirred conditions (with stirring bar present) some vials were still without crystals even after period III (24 hours). Interestingly, there was a stark difference in polymorphic outcomes between solutions stirred at 100 or 1600 rpm (Fig. 5e–h) and the unstirred solutions with the stirrer present at 0 rpm (Fig. 5c and d). For vials where no crystals were visible at the end of the cooling (period I), once stirring was turned on, α crystallised overwhelmingly, rather than γ predominantly found in the unstirred solutions. At 100 rpm, almost all vials crystallised as pure α when stirred at 0 °C, whereas at 25 °C approximately half of the vials crystallised as α + γ mixtures at higher concentrations (Fig. 5e and f). At 1600 rpm, pure α was found at both 0 °C and 25 °C (Fig. 5g and h), which shows that more vigorous agitation promotes crystallisation of α.
Table 1 summarises the combined results over all concentrations for the eight different experimental conditions shown in Fig. 5. For unstirred experiments with the stirring bar present (0 rpm), of the samples that crystallised during the isothermal period II, pure γ was found in 98% of 61 samples (0 °C) and 73% of 37 samples (25 °C). Remarkably, all four stirring experiments showed that the α form was clearly preferred over the γ form. When stirring was used (isothermal period II), of the samples that crystallised, pure α was found in 98% of 60 samples (0 °C, 100 rpm), 72% of 76 samples (25 °C, 100 rpm), 99% of 76 samples (0 °C, 1600 rpm), and 99% of 84 samples (25 °C, 1600 rpm). For experiments at 25 °C and 100 rpm where only 72% were α, the remaining samples were a mixture of α and γ. Furthermore, we note that during the cooling period I, when no stirring was used, γ crystallised predominantly.
Our observations are consistent with those of Devi et al. who previously investigated the effect of stirring rate and concentration on the crystallisation of glycine upon rapid cooling.11 Our experimental conditions in Fig. 5e correspond to Devi et al.'s experiments at relative supersaturations between 1.10 to 1.30 in their nucleation matrix and with similar stirring rates. Under these conditions, Devi et al. obtained pure α glycine, which is in agreement with our results. We note that Devi et al. performed one experiment at each point of their nucleation matrix. In our set of experiments, of the 60 samples that nucleated, only one produced a mixture of α and γ, and so it is unsurprising that Devi et al. did not obtain any γ in experiments they performed under corresponding conditions.
Our results show that stirring promotes formation of the metastable α polymorph which could be due to primary nucleation of a metastable polymorph (i.e. either α or β, which is quickly replaced by α), or due to secondary nucleation, induced by presence of γ crystals which nucleate in the absence of agitation, and especially so in the presence of the PTFE coated stirrer bar. In the case of secondary nucleation, we would expect to see a mixture of α and γ, as γ (the stable form) would always be present, and 28% of samples that crystallised in period II of experiment shown in Fig. 5f were α and γ mixtures. Since γ is known to grow more slowly than α, it is also possible that in the competition between α and γ polymorphs growing concurrently the fraction of γ in the resulting solid glycine powder is too low to be detectable.
However, the secondary nucleation mechanism would not fully explain our observations, since γ did not crystallise in many vials at lower concentrations at 25 °C and thus no crystals would be present to induce nucleation in the large majority of vials, all of which then crystallised upon stirring. Note that crystal growth rates for both α and γ are of the order of at least 1 micron per second at these concentrations so if any crystal nucleates it would be several millimetres long within an hour.42 Our results indicate that primary nucleation of metastable polymorphs of glycine is strongly enhanced by stirring, in agreement with our previous observations from controlled shear experiments.34 While molecular scale effects are not likely to be affected by macroscopic stirring, colloidal scale precursor clusters have been implicated as a possible explanation why stirring favours primary nucleation of α glycine.41
Results of our glycine crystallisation experiments under stirring conditions clearly show that polymorphic outcomes can be controlled by agitation. While enhancement of either primary or secondary nucleation of specific polymorphs may be responsible for effects of agitation on polymorphic crystallisation, in the case of glycine investigated here there appears to be the clear effects of stirring on primary nucleation of α glycine.
There is a general point to be made here, which is relevant not only to glycine but to many other systems where stirring and interfacial effects may be determining nucleation kinetics and polymorphic outcomes, that in order to relate experimental observations to possible nucleation mechanisms it is necessary to consider and control these effects in order to separate them from those commonly investigated as drivers for nucleation control, such as solvent, concentration and temperature.
4. Conclusions
Polymorphic crystallisation of glycine from highly concentrated aqueous solutions under isothermal conditions was found to be highly sensitive to stirring. Under quiescent conditions, crystallisation resulted in preferential formation of γ glycine. This shows that γ glycine can be crystallised from purely aqueous solutions in glass vials, using highly concentrated glycine concentrations prepared at high temperature and cooling them to low crystallisation temperature without agitation. This is contrary to a commonly held view that γ glycine does not form upon cooling from pure aqueous solutions without any additives, co-solvent or external fields, which is due to the fact that typical cooling crystallisation experiments are performed at lower concentrations than we used here and in the presence of agitation.The presence of a PTFE-coated magnetic stirring bar (without stirring) significantly enhanced the crystallisation rate but did not significantly change the polymorphic outcome. The effect of stirring was twofold. First, the crystallisation rate further increased so that all vials nucleated by the end of the stirred isothermal period, and, second, stirring resulted in preferential formation of α glycine. This is surprising and while secondary nucleation might be a contributing factor, we believe that it is primarily due to enhancement of primary nucleation of α glycine by agitation.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank EPSRC and the Future Continuous Manufacturing and Advanced Crystallisation Research Hub (Grant Ref: EP/P006965/1) for funding this work. The authors would like to acknowledge that this work was carried out in the CMAC National Facility supported by UKRPIF (UK Research Partnership Fund) award from the Higher Education Funding Council for England (HEFCE) (Grant ref HH13054).References
- A. Llinàs and J. M. Goodman, Drug Discovery Today, 2008, 13, 198–210 CrossRef PubMed.
- J. Lu, Z. Li and X. Jiang, Front. Chem. Eng. China, 2010, 4, 37–44 CrossRef CAS.
- H. G. Brittain, Polymorphism in Pharmaceutical Solids, Informa Healthcare, New York, 2nd edn, 2009 Search PubMed.
- F. Artusio and R. Pisano, Int. J. Pharm., 2018, 547, 190–208 CrossRef CAS PubMed.
- J. W. Chew, S. N. Black, S. P. Chow, R. B. H. Tan and K. J. Carpenter, CrystEngComm, 2007, 1, 128–130 RSC.
- E. A. Losev, M. A. Mikhailenko, A. F. Achkasov and E. V. Boldyreva, New J. Chem., 2013, 37, 1973–1981 RSC.
- J. Liu, M. Svärd and A. C. Rasmuson, Cryst. Growth Des., 2014, 14, 5521–5531 CrossRef CAS.
- K. Sypek, I. S. Burns, A. J. Florence and J. Sefcik, Cryst. Growth Des., 2012, 12, 4821–4828 CrossRef CAS.
- C. Cashell, D. Corcoran and B. K. Hodnett, J. Cryst. Growth, 2004, 273, 258–265 CrossRef CAS.
- Y. Tahri, E. Gagnière, E. Chabanon, T. Bounahmidi and D. Mangin, J. Cryst. Growth, 2016, 435, 98–104 CrossRef CAS.
- K. Renuka Devi, V. Gnanakamatchi and K. Srinivasan, J. Cryst. Growth, 2014, 400, 34–42 CrossRef CAS.
- A. L. Markel, A. F. Achkasov, T. A. Alekhina, O. I. Prokudina, M. A. Ryazanova, T. N. Ukolova, V. M. Efimov, E. V. Boldyreva and V. V. Boldyrev, Pharmacol., Biochem. Behav., 2011, 98, 234–240 CrossRef CAS PubMed.
- A. Jawor-Baczynska, B. D. Moore, H. S. Lee, A. V. McCormick and J. Sefcik, Faraday Discuss., 2013, 167, 425–440 RSC.
- L. Fábián and A. Kálmán, Acta Crystallogr., Sect. B: Struct. Sci., 2004, 60, 547–558 CrossRef PubMed.
- E. V. Boldyreva, V. A. Drebushchak, T. N. Drebushchak, I. E. Paukov, Y. A. Kovalevskaya and E. S. Shutova, J. Therm. Anal. Calorim., 2003, 73, 419–428 CrossRef CAS.
- Y. Iitaka, Nature, 1959, 183, 390–391 CrossRef CAS.
- C. S. Towler, R. J. Davey, R. W. Lancaster and C. J. Price, J. Am. Chem. Soc., 2004, 126, 13347–13353 CrossRef CAS PubMed.
- S. A. Rivera, D. G. Allis and B. S. Hudson, Cryst. Growth Des., 2008, 8, 3905–3907 CrossRef CAS.
- G. L. Perlovich, L. K. Hansen and A. Bauer-Brandl, J. Therm. Anal. Calorim., 2001, 66, 699–715 CrossRef CAS.
- Y. Liu, M. H. van den Berg and A. J. Alexander, Phys. Chem. Chem. Phys., 2017, 19, 19386–19392 RSC.
- X. Yang, X. Wang and C. B. Ching, J. Chem. Eng. Data, 2008, 53, 1133–1137 CrossRef CAS.
- K. Park, J. M. B. Evans and A. S. Myerson, Cryst. Growth Des., 2003, 3, 991–995 CrossRef CAS.
- L. Dang, H. Yang, S. Black and H. Wei, Org. Process Res. Dev., 2009, 13, 1301–1306 CrossRef CAS.
- X. Yang, J. Lu, X. J. Wang and C. B. Ching, J. Cryst. Growth, 2008, 310, 604–611 CrossRef CAS.
- Y. Cui and A. S. Myerson, Cryst. Growth Des., 2014, 14, 5152–5157 CrossRef CAS.
- R. Dowling, R. J. Davey, R. A. Curtis, G. Han, S. K. Poornachary, S. Chow and R. B. H. Tan, Chem. Commun., 2010, 46, 5924–5926 RSC.
- Q. Jiang, A. G. Shtukenberg, M. D. Ward and C. Hu, Cryst. Growth Des., 2015, 15, 2568–2573 CrossRef CAS.
- K. Renuka Devi and K. Srinivasan, J. Cryst. Growth, 2014, 401, 227–232 CrossRef CAS.
- I. Weissbuch, R. Popovitz-Biro, M. Lahav and L. Leiserowitz, Acta Crystallogr., Sect. B: Struct. Sci., 1995, 51, 115–148 CrossRef.
- Y. Iitaka, Acta Crystallogr., 1961, 14, 1–14 CrossRef CAS.
- G. He, V. Bhamidi, S. R. Wilson, R. B. H. Tan, P. J. A. Kenis and C. F. Zukoski, Cryst. Growth Des., 2006, 6, 1746–1749 CrossRef CAS.
- M. Xu and K. D. M. Harris, J. Phys. Chem. B, 2007, 111, 8705–8707 CrossRef CAS PubMed.
- N. V. Surovtsev, S. V. Adichtchev, V. K. Malinovsky, A. G. Ogienko, V. A. Drebushchak, A. Y. Manakov, A. I. Ancharov, A. S. Yunoshev and E. V. Boldyreva, J. Chem. Phys., 2012, 137, 65103 CrossRef CAS PubMed.
- C. Forsyth, P. A. Mulheran, C. Forsyth, M. D. Haw, I. S. Burns and J. Sefcik, Cryst. Growth Des., 2015, 15, 94–102 CrossRef CAS.
- K. Igarashi, Y. Sasaki, M. Azuma, H. Noda and H. Ooshima, Eng. Life Sci., 2003, 3, 159–163 CrossRef CAS.
- G. Han, S. K. Poornachary, P. S. Chow and R. B. H. Tan, Cryst. Growth Des., 2010, 10, 4883–4889 CrossRef CAS.
- N. Javid, T. Kendall, I. S. Burns and J. Sefcik, Cryst. Growth Des., 2016, 16, 4196–4202 CrossRef CAS.
- X. Sun, B. A. Garetz and A. S. Myerson, Cryst. Growth Des., 2006, 6, 684 CrossRef CAS.
- M. Kuhs, J. Zeglinski and Å. C. Rasmuson, Cryst. Growth Des., 2014, 14, 905–915 CrossRef CAS.
- F. L. Nordström, M. Svärd, B. Malmberg and Å. C. Rasmuson, Cryst. Growth Des., 2012, 12, 4340–4348 CrossRef.
- C. Forsyth, I. S. Burns, P. A. Mulheran and J. Sefcik, Cryst. Growth Des., 2016, 16, 136–144 CrossRef CAS.
- G. Han, P. S. Chow and R. B. H. Tan, Cryst. Growth Des., 2012, 12, 2213–2220 CrossRef CAS.
Footnotes |
| † Raw data is available open access through the following DOI: https://doi.org/10.15129/9a4ec7f5-fe41-4ec2-92ef-a9abca03b217 |
| ‡ Electronic supplementary information (ESI) available: Contains information about quenched cooling experiments and movement induced nucleation. See DOI: 10.1039/c8ce01829d |
| This journal is © The Royal Society of Chemistry 2019 |