
A phase-transfer crystallization pathway to synthesize ultrasmall silicoaluminophosphate for enhanced catalytic conversion of dimethylether-to-olefin†

Abstract
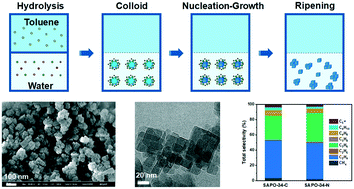
We present a phase-transfer crystallization approach to the synthesis of ultrasmall (<100 nm) SAPO-34 crystallites. This method features porogen-free and tumbling crystallization in biphasic toluene–water media. The nanosized SAPO-34 catalyst has demonstrated prolonged catalysis lifetime (approximately 5 fold) and higher ethylene/propylene selectivity (by ca. 5%) for dimethylether-to-olefin conversion.
 Please wait while we load your content...
Please wait while we load your content...

