
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Reversible tricolour luminescence switching based on a piezochromic iridium(III) complex†
Tianzhi
Yang
a,
Yue
Wang
a,
Xingman
Liu
a,
Guangfu
Li
a,
Weilong
Che
a,
Dongxia
Zhu
*a,
Zhongmin
Su
 *a and
Martin R.
Bryce
*b
*a and
Martin R.
Bryce
*b
aKey Laboratory of Nanobiosensing and Nanobioanalysis at Universities of Jilin Province, Department of Chemistry, Northeast Normal University, 5268 Renmin Street, Changchun, Jilin Province 130024, P. R. China. E-mail: zhudx047@nenu.edu.cn; zmsu@nenu.edu.cn
bDepartment of Chemistry, Durham University, Durham, DH1 3LE, UK. E-mail: m.r.bryce@durham.ac.uk
First published on 21st November 2019
Abstract
On the basis of rational molecular design, the tricolour luminescence switching of an Ir(III) complex is achieved for the first time. The transformation between two crystalline states and an amorphous state is responsible for the switching behaviour of this complex between blue, green and yellow states. Solvent molecules are shown to play a crucial role in the crystallization and luminescence processes.
Piezochromic luminescent (PCL) materials, the emission properties of which are responsive to mechanical stimuli, have captured extensive attention in the construction of mechano-sensors, security inks, optical data recording and storage devices.1 A variety of PCL materials have been reported to date, but most of them only display reversible bicolour switching.2 PCL materials possessing multiple luminescent colours remain rare due to the lack of universal design principles.3 The polymorphs of organic dyes usually show different emission properties because of their dissimilar molecular arrangements,4 and thus, utilizing the transformation between them should be a promising approach to achieve multicolour luminescence switching.5 Compared to bicolour switching materials, multicolour counterparts have enhanced colour contrast and patterning, which is beneficial for the practical applications noted above. Reported multicolour examples include small organic molecules,6 liquid crystals,7 amphiphilic molecules,8 multi-component assemblies,9 polymers,10 gold(I)11 and platinum(II)12 complexes.
Nowadays, Ir(III) complexes are established as an excellent class of phosphorescent emitters for various optoelectronic applications.13 Several Ir(III) complexes, including the classical fac-Ir(ppy)3, have been shown to be polymorphic.14 However, it appears that no experimental results are available for the multicolour luminescence switching based on Ir(III) complexes. Therefore, it is of great significance to explore whether Ir(III) complexes can provide a new family of multicolour PCL materials.
To address this issue, a novel cationic Ir(III) complex [Ir(Mesptz)2Mtzpy]+PF6−, (“complex 1” hereafter), in which Mesptz is 1-mesityl-5-phenyl-1H-1,2,4-triazole and Mtzpy is 2-(1-methyl-1H-tetrazol-5-yl)pyridine, was rationally designed on the basis of the following considerations. First, the bulky mesityl group on the ligands should increase the intermolecular distances thereby inducing a loose crystal structure,15 which is more prone to collapse into an amorphous state under external pressure.16 In general, the transition toward an amorphous phase might alter the emission of crystalline luminophores, resulting in PCL behaviour.2 Second, previous studies indicate that polymorphism is more favoured in heterocyclic compounds, perhaps because the interactions caused by heteroatoms, such as C–H⋯N and C–H⋯O hydrogen bonds, provide more opportunity to form different crystal structures.17 Therefore, triazolyl and tetrazolyl groups were introduced into complex 1 to facilitate the formation of polymorphs. Third, as recognized by a number of studies on the aggregation-induced emission (AIE) or aggregation-enhanced emission (AEE) phenomena, the restriction of intramolecular rotations and/or vibrations usually leads to a remarkable emission enhancement.18 Accordingly, relatively rigid ligands were employed to weaken the intramolecular motions,19 and thereby to endow complex 1 with an intense phosphorescence.
As a result of these combined design features, three powder samples of complex 1 with different emission colours, blue, green and yellow, were observed. These three samples (hereafter referred to as B-form, G-form and Y-form, respectively) can be switched repeatedly by mechanical stimulation or solvent treatment. Moreover, their highly efficient solid-state emissions are conducive to practical applications, and a simple proof-of-concept tricolour optical recording device was constructed using this complex as the active material. To the best of our knowledge, this is the first Ir(III) complex that displays tricolour luminescence switching.
The target complex 1 was prepared in high yield following a previous method (see the Scheme S1, ESI†). Complex 1 is weakly luminescent in dilute acetonitrile solution but displays a strikingly enhanced emission in acetonitrile–water mixture with a 90 vol% water fraction, confirming that complex 1 is AEE-active (Fig. 1b).18 Transmission electron microscopy and electron diffraction experiments demonstrated that amorphous aggregates of complex 1 are formed in the mixtures with high water contents (Fig. S2a and b, ESI†).20 Furthermore, rigidifying the molecules by doping complex 1 into PMMA matrices leads to a significant enhancement of emission intensity even at a very low doping concentration of 0.05 wt% (Fig. S2c, ESI†). Therefore, we tentatively ascribe the mechanism of AEE to the restriction of intramolecular rotations and vibrations.21
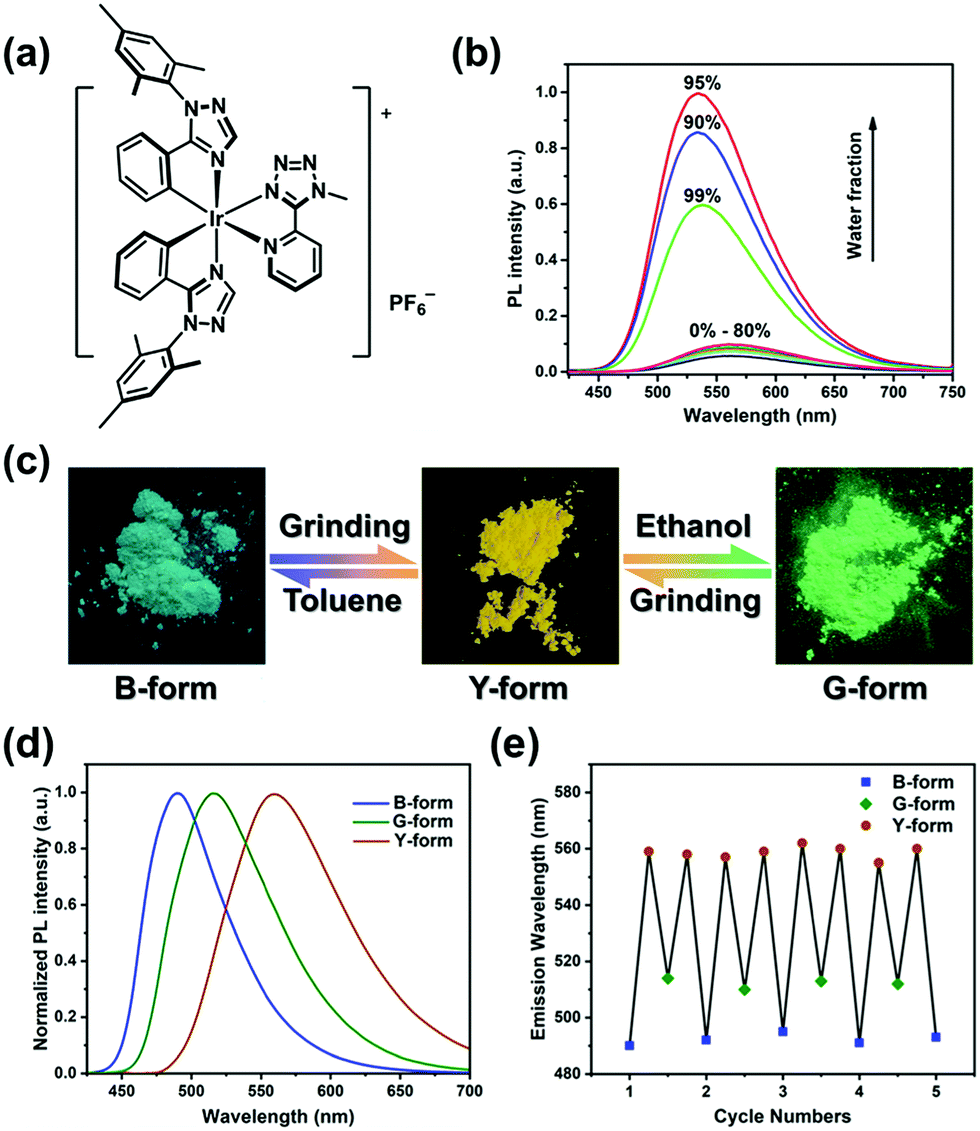 | ||
| Fig. 1 (a) Chemical structure of complex 1. (b) Normalized emission spectra of complex 1 in CH3CN–H2O mixtures (complex concentration = 5.0 × 10−5 M) with different water fractions (0–99% v/v) at room temperature. (c) Photographs and schematic representation of a tricolour luminescence switching process. (d) Normalized emission spectra of B-, G- and Y-forms. (e) Repeated cycles of the tricolour luminescence switching process. | ||
The B-form and G-form, two original powder samples, were obtained by recrystallization of complex 1 from the mixed solvents of toluene–dichloromethane and ethanol–acetonitrile, respectively. Under UV-light irradiation, the B-form exhibited a bluish-green emission peak with the maximum (λmax) at 492 nm, whereas the G-form emitted green phosphorescence (λmax = 515 nm) (Fig. 1d). Both of the samples can be transformed into a yellow-emitting powder (Y-form, λmax = 559 nm) upon mechanical grinding with hand pressure in a mortar, demonstrating their PCL characteristics (Fig. 1c and d). After wetting Y-form with toluene, the emission colour reverted rapidly to bluish-green. Correspondingly, the emission of Y-form can be returned to green by the dropwise addition of ethanol, in a few seconds. It should be noted that complex 1 is slightly soluble in toluene and ethanol. In addition, when the solvent-treated samples were further ground, a red-shifted emission reappeared again. This tricolour switching process is highly reversible for several cycles (Fig. 1e). The photophysical characteristics of the B-, G-, and Y-forms are summarized in Table 1.
| λ em (nm) | Φ em | τ (μs) | k r × 106 s−1 | k nr × 106 s−1 | |
|---|---|---|---|---|---|
| a λ ex = 365 nm. b Measured in CH3CN (1.0 × 10−5 M); λabs [nm] (ε [×104 M−1 cm−1]) 252 (0.98), 319 (0.25), 355 (0.22). | |||||
| B-form | 492 | 0.793 | 0.691 | 1.15 | 0.30 |
| G-form | 515 | 0.682 | 0.518 | 1.32 | 0.61 |
| Y-form | 559 | 0.568 | 0.592 | 0.96 | 0.73 |
| Solutionb | 566 | 0.074 | 0.065 | 1.13 | 14.2 |
As illustrated in Fig. S3 (ESI†), the similar 1H NMR spectra of all three samples (B-, G- and Y-forms) imply that their tricolour switching behaviour is a physical process rather than a chemical reaction.22 Hence, to understand the origin of this process, the powder X-ray diffraction (PXRD) patterns of the B-, G- and Y-forms were obtained. The B- and G-forms displayed several intense and sharp reflection peaks, indicating their well-ordered crystalline nature (Fig. 2a). In contrast, these diffraction peaks disappeared or became weaker and broader after grinding, suggesting that the Y-form is a disordered amorphous phase. By treating Y-form with toluene or ethanol, the diffraction peaks similar to the those of B-form or G-form appeared again, which confirmed that their crystalline structures were recovered. Consequently, the red-shift of the emission triggered by grinding can be ascribed to a crystalline-to-amorphous phase transition.
 | ||
| Fig. 2 (a) PXRD patterns of the corresponding powder samples. (b) The molecular conformational comparison of B-crystal and G-crystal. (c) The diagrams and energy levels of molecular orbitals as well as crucial electronic excitation processes for B-form and G-form. | ||
The diverse PXRD patterns of B- and G-form suggest that their different emission properties are associated with their molecular arrangements. In order to verify this proposition, single crystals (B-crystal and G-crystal) suitable for X-ray structural analysis were obtained by slow evaporation from a saturated toluene solution, or by cooling a hot ethanol solution. The simulated diffraction peaks of B-crystal and G-crystal are consistent with the PXRD data of B-form and G-form, respectively, manifesting their similar crystal packing (Fig. 2a). Both B- and G-crystal belong to the monoclinic system and crystallize in the P21/c space groups. The molecules adopt antiparallel coupling in B-crystal; whereas in G-crystal, each pair of molecules packs in a herringbone fashion as shown in Fig. S4a (ESI†). As expected, their molecular packing is very loose and therefore can be easily modified by the external pressure, leading to the aforementioned PCL behaviour.
There is a noteworthy distinction between B- and G-crystal in the molecular conformation, especially in the extent of ligand distortion (Fig. 2b). In terms of cyclometalated ligands, the dihedral angles between the phenyl and triazolyl groups in B-crystal are 5.30° and 5.46°, which are smaller than the values of 8.53° and 7.83° in G-crystal. Besides, the Mtzpy N^N ligand is more twisted in B-crystal than in G-crystal, with the dihedral angle being 9.10° in the former and 6.95° in the latter (Fig. S4b, ESI†).
To probe the effect of molecular conformation on the emission characteristics, quantum chemical calculations were performed. The molecular orbitals involved in the excitation, as well as the important electronic transitions, are depicted in Fig. 2c. According to the calculated results, the distributions of the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) are similar in both the B-form and G-form. As expected, the mesityl groups of the cyclometalated ligands do not contribute to the frontier orbitals.15 The HOMOs mainly reside on the iridium centre and the phenyltriazole fragments of the cyclometalated C^N ligands, while the LUMOs primarily localize on the N^N ligand, therefore the Mtzpy ligand is not considered to be ancillary. Therefore, the molecular orbital energy levels of 1 should be strongly affected by the conformation of relevant ligands. Although the structural differences between B- and G- forms are small (Fig. 2b), it is considered that the more planar phenyltriazole fragments help to stabilize the HOMO energy level of B-form. Meanwhile, the more twisted Mtzpy ligand in B-form gives rise to its higher LUMO energy level. As a consequence, the molecular conformation of B-form would lead to a larger HOMO–LUMO energy gap. Interestingly, time-dependent density functional theory (TD-DFT) calculations establish that the T1 state of complex 1 mainly originates from the excitation of HOMO → LUMO (92.2% and 87.7% for B-form and G-form, respectively). Hence, the emission of B-form is apparently blue-shifted compared to that of G-form.
In addition, the intermolecular interactions caused by heteroatoms were observed, as anticipated. However, we had not predicted that multiple interactions related to solvent molecules would exist in these two crystals (Fig. 3a). In the B-crystal, three kinds of C–H⋯π hydrogen bonds (interaction I, II and III) are formed between complex 1 and toluene molecules with the interaction distances of 2.81 Å, 2.43 Å and 2.81 Å, and the interaction angles of 154.0°, 135.8° and 135.4°, respectively. In the case of the G-crystal, there are multiple interactions. These are between the ligands of complex 1 (C–H⋯N IV, 2.47 Å, 152.2°), and involving the PF6− and ethanol molecules through C–H⋯F (V, 2.50 Å, 175.2°), O–H⋯F (VI, 2.17 Å, 159.4°), and C–H⋯π (VII, 3.09 Å, 126.7°) interactions. Thus, it is rational to propose that solvent molecules also participate strongly in the crystallization process.
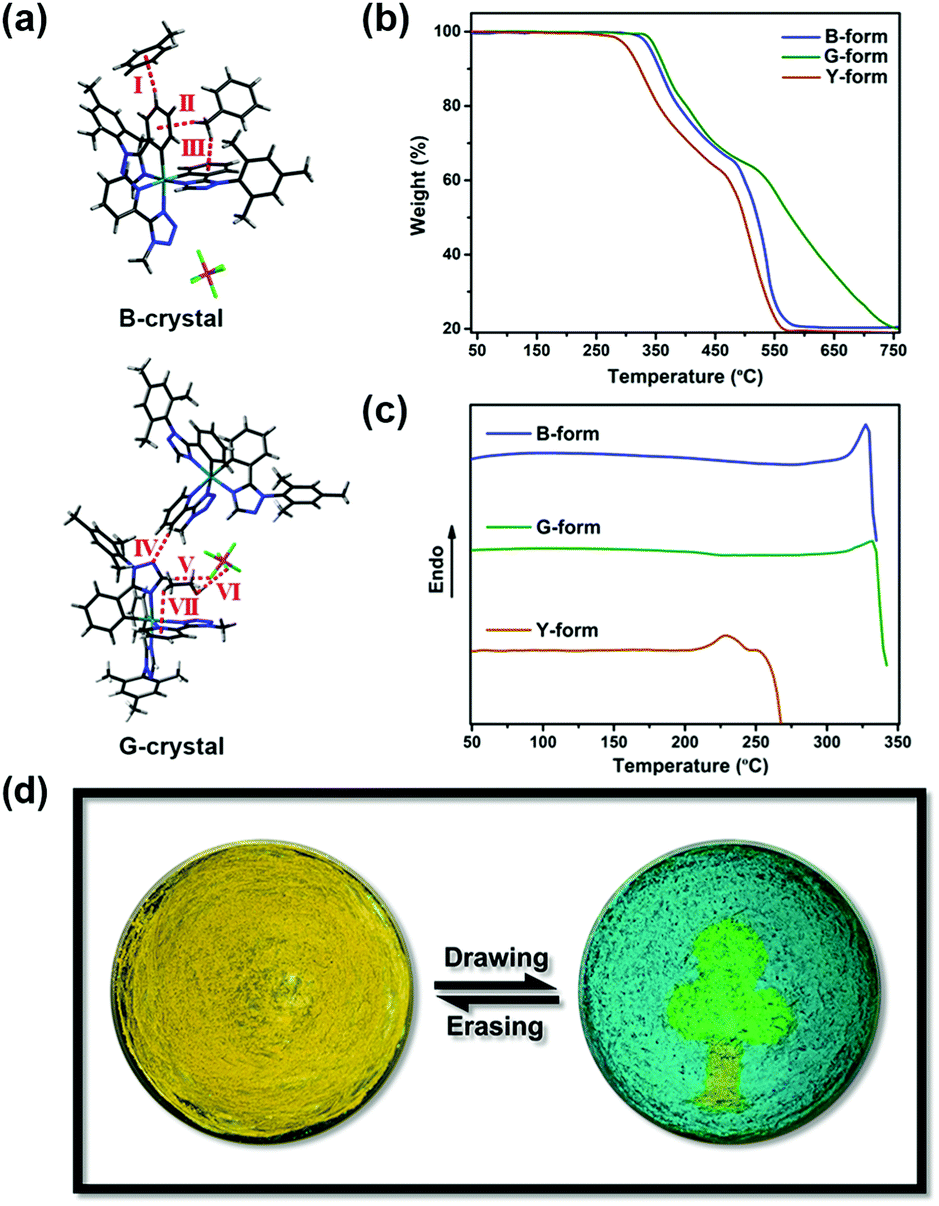 | ||
| Fig. 3 (a) Intermolecular interactions in the B-crystal and G-crystal. (b and c) TGA graphs and DSC traces of B-, G- and Y-forms. (d) Photographs and schematic representation of a patterned optical recording system. | ||
Given the different molecular arrangements and conformations of B-, G- and Y-forms, the thermal stability of these three samples was evaluated by thermogravimetric analysis (TGA). The decomposition temperature of Y-form is lower than that of both B- and G-forms, which should be attributed to the amorphous nature of Y-form (Fig. 3b). Moreover, there are no endothermic or exothermic peaks prior to the decomposition temperature in the differential scanning calorimetry (DSC) curves of B-form or G-form (Fig. 3c). Upon heating Y-form to ca. 220 °C, the DSC curve only shows an endothermic melting point peak due to different molecular aggregation states.23 Indeed, without the indispensable solvent molecules, the transformation among the different forms cannot be achieved by thermal treatment, which further validates the essential role of solvent molecules.
By exploiting the tricolour switching property of complex 1, a simple patterned optical recording system visualized as a tree was fabricated in an agate mortar (Fig. 3d). The procedure was as follows: first, Y-form was spread evenly on the mortar, exhibiting a yellow emission under photoexcitation. Then, a green-emitting crown and a blue-emitting sky were drawn by using a glass capillary with ethanol or toluene. A part of the Y-form sample was retained as a tree-trunk. Finally, this pattern could be erased by mechanical grinding with hand pressure, recovering the original yellow background. This drawing and erasing process was repeated for many cycles without any obvious performance degradation.
In summary, tricolour luminescence switching has been observed in an Ir(III) complex for the first time. Complex 1 can form two different crystal structures, and the transformation between two crystalline states and an amorphous state leads to tricolour switching behaviour. Solvent molecules are shown to play a crucial role in the crystallization and luminescence processes. It is proposed that the different emission characteristics of B-form and G-form should be ascribed to their distinctions in molecular arrangement, conformation and intermolecular interactions. The clear and reversible tricolour luminescence switching feature makes complex 1 a competitive candidate for practical applications. More importantly, it is envisioned that our molecular design strategies will provide valuable guidelines for developing ideal multicolour switching luminescent materials in the future.
This work was funded by NSFC (no. 51473028), the Key Scientific and Technological Project of Jilin Province (20190701010GH), and the Development and Reform Commission of Jilin Province (20160058). M. R. B. thanks EPSRC grant EL/L02621X/1 for funding.
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) Y. Liu, Q. Zeng, B. Zou, Y. Liu, B. Xu and W. Tian, Angew. Chem., Int. Ed., 2018, 57, 15670 CrossRef CAS PubMed; (b) S. K. Park, I. Cho, J. Gierschner, J. H. Kim, J. H. Kim, J. E. Kwon, O. K. Kwon, D. R. Whang, J.-H. Park, B.-K. An and S. Y. Park, Angew. Chem., Int. Ed., 2016, 55, 203 CrossRef CAS PubMed; (c) Y. Wang, X. Tan, Y.-M. Zhang, S. Zhu, I. Zhang, B. Yu, K. Wang, B. Yang, M. Li, B. Zou and S. X.-A. Zhang, J. Am. Chem. Soc., 2015, 137, 931 CrossRef CAS PubMed.
- (a) Y. Sagara, S. Yamane, M. Mitani, C. Weder and T. Kato, Adv. Mater., 2016, 28, 1073 CrossRef CAS PubMed; (b) Z. Chi, X. Zhang, B. Xu, X. Zhou, C. Ma, Y. Zhang, S. Liu and J. Xu, Chem. Soc. Rev., 2012, 41, 3878 RSC.
- Z. Ma, Z. Wang, M. Teng, Z. Xu and X. Jia, ChemPhysChem, 2015, 16, 1811 CrossRef CAS PubMed.
- (a) H. Liu, Y. Shen, Y. Yan, C. Zhou, S. Zhang, B. Li, L. Ye and B. Yang, Adv. Funct. Mater., 2019, 29, 1901895 CrossRef; (b) B. Huang, W.-C. Chen, Z. Li, J. Zhang, W. Zhao, Y. Feng, B. Z. Tang and C.-S. Lee, Angew. Chem., Int. Ed., 2018, 57, 12473 CrossRef CAS PubMed.
- (a) Y. Zhang, Q. Song, K. Wang, W. Mao, F. Cao, J. Sun, L. Zhan, Y. Lv, Y. Ma, B. Zou and C. Zhang, J. Mater. Chem. C, 2015, 3, 3049 RSC; (b) Z. He, L. Zhang, J. Mei, T. Zhang, J. W. Y. Lam, Z. Shuai, Y. Q. Dong and B. Z. Tang, Chem. Mater., 2015, 27, 6601 CrossRef CAS.
- (a) M. Okazaki, Y. Takeda, P. Data, P. Pander, H. Higginbotham, A. P. Monkman and S. Minakata, Chem. Sci., 2017, 8, 2677 RSC; (b) Z. Ma, Z. Wang, X. Meng, Z. Ma, Z. Xu, Y. Ma and X. Jia, Angew. Chem., Int. Ed., 2016, 55, 519 CrossRef CAS PubMed.
- Y. Sagara and T. Kato, Angew. Chem., Int. Ed., 2011, 50, 9128 CrossRef CAS PubMed.
- S. Yagai, S. Okamura, Y. Nakano, M. Yamauchi, K. Kishikawa, T. Karatsu, A. Kitamura, A. Ueno, D. Kuzuhara, H. Yamada, T. Seki and H. Ito, Nat. Commun., 2014, 5, 4013 CrossRef CAS PubMed.
- (a) H.-J. Kim, D. R. Whang, J. Gierschner, C. H. Lee and S. Y. Park, Angew. Chem., Int. Ed., 2015, 54, 4330 CrossRef CAS PubMed; (b) Y. Matsunaga and J.-S. Yang, Angew. Chem., Int. Ed., 2015, 54, 7985 CrossRef CAS PubMed.
- A. Lavrenova, D. W. R. Balkenende, Y. Sagara, S. Schrettl, Y. C. Simon and C. Weder, J. Am. Chem. Soc., 2017, 139, 4302 CrossRef CAS PubMed.
- T. Seki, T. Ozaki, T. Okura, K. Asakura, A. Sakon, H. Uekusa and H. Ito, Chem. Sci., 2015, 6, 2187 RSC.
- S. J. Choi, J. Kuwabara, Y. Nishimura, T. Arai and T. Kanbara, Chem. Lett., 2012, 41, 65 CrossRef CAS.
- Iridium(III) in Optoelectronic and Photonics Applications, ed. E. Zysman-Colman, J. Wiley & Sons, Ltd, 2017 Search PubMed.
- (a) H. Benjamin, Y. Zheng, A. S. Batsanov, M. A. Fox, H. A. Al-Attar, A. P. Monkman and M. R. Bryce, Inorg. Chem., 2016, 55, 8612 CrossRef CAS PubMed; (b) S. Takayasu, T. Suzuki and K. Shinozaki, J. Phys. Chem. B, 2013, 117, 9449 CrossRef CAS PubMed; (c) T. F. Mastropietro, Y. J. Yadav, E. I. Szerb, A. M. Talarico, M. Ghedini and A. Crispini, Dalton Trans., 2012, 41, 8899 RSC.
- (a) A. F. Henwood, D. Antón-García, M. Morin, D. Rota Martir, D. B. Cordes, C. Casey, A. M. Z. Slawin, T. Lebl, M. Bühl and E. Zysman-Colman, Dalton Trans., 2019, 48, 9639 RSC; (b) D. G. Congrave, A. S. Batsanov and M. R. Bryce, Dalton Trans., 2018, 47, 16524 RSC; (c) W. Zhang, J. Ma, G.-J. Liu, X.-Y. Liu, J. Fan and L.-S. Liao, J. Mater. Chem. C, 2017, 5, 9496 RSC.
- G. Li, X. Ren, G. Shan, W. Che, D. Zhu, L. Yan, Z. Su and M. R. Bryce, Chem. Commun., 2015, 51, 13036 RSC.
- (a) Y. Dong, B. Xu, J. Zhang, X. Tan, L. Wang, J. Chen, H. Lv, S. Wen, B. Li, L. Ye, B. Zou and W. Tian, Angew. Chem., Int. Ed., 2012, 51, 10782 CrossRef CAS PubMed; (b) Y. Dong, J. Zhang, X. Tan, L. Wang, J. Chen, B. Li, L. Ye, B. Xu, B. Zou and W. Tian, J. Mater. Chem. C, 2013, 1, 7554 RSC; (c) H. Y. Zhang, Z. L. Zhang, K. Q. Ye, J. Y. Zhang and Y. Wang, Adv. Mater., 2006, 18, 2369 CrossRef CAS.
- J. Mei, N. L. C. Leung, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Chem. Rev., 2015, 115, 11718 CrossRef CAS PubMed.
- D. Ma, T. Tsuboi, Y. Qiu and L. Duan, Adv. Mater., 2017, 29, 1603253 CrossRef PubMed.
- Y. Jiang, G. Li, W. Che, Y. Liu, B. Xu, G. Shan, D. Zhu, Z. Su and M. R. Bryce, Chem. Commun., 2017, 53, 3022 RSC.
- Y. Wu, H.-Z. Sun, H.-T. Cao, H.-B. Li, G.-G. Shan, Y.-A. Duan, Y. Geng, Z.-M. Su and Y. Liao, Chem. Commun., 2014, 50, 10986 RSC.
- Y. Wang, T. Yang, X. Liu, G. Li, W. Che, D. Zhu and Z. Su, J. Mater. Chem. C, 2018, 6, 12217 RSC.
- J. Bernstein, Polymorphism in Molecular Crystals, Oxford University Press, New York, 2002 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, figures and procedures for the DFT calculations. CCDC 1864016 and 1864017. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9cc08545a |
| This journal is © The Royal Society of Chemistry 2019 |