
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA stapled chromogranin A-derived peptide is a potent dual ligand for integrins αvβ6 and αvβ8†
Francesca
Nardelli‡
 a,
Michela
Ghitti
*a,
Giacomo
Quilici
a,
Alessandro
Gori
b,
Qingqiong
Luo
c,
Andrea
Berardi
a,
Angelina
Sacchi
a,
Matteo
Monieri
a,
Greta
Bergamaschi
b,
Wolfgang
Bermel
d,
Fuxiang
Chen
c,
Angelo
Corti
*ae,
Flavio
Curnis
*a and
Giovanna
Musco
*a
a,
Michela
Ghitti
*a,
Giacomo
Quilici
a,
Alessandro
Gori
b,
Qingqiong
Luo
c,
Andrea
Berardi
a,
Angelina
Sacchi
a,
Matteo
Monieri
a,
Greta
Bergamaschi
b,
Wolfgang
Bermel
d,
Fuxiang
Chen
c,
Angelo
Corti
*ae,
Flavio
Curnis
*a and
Giovanna
Musco
*a
aIRCCS San Raffaele Scientific Institute, Via Olgettina 58, 20132 Milan, Italy. E-mail: corti.angelo@hsr.it; curnis.flavio@hsr.it; ghitti.michela@hsr.it; musco.giovanna@hsr.it
bIstituto di Chimica del Riconoscimento Molecolare, C.N.R., Via Mario Bianco 9, 20131 Milan, Italy
cNinth People's Hospital, Shanghai Jiao Tong University School of Medicine, 639 Zhizaoju Road, Shanghai, 200011, China
dBruker BioSpin GmbH, Silberstreifen 4, Rheinstetten, 76287, Germany
eVita Salute San Raffaele University, Via Olgettina 58, 20132 Milan, Italy
First published on 22nd November 2019
Abstract
Combining 2D STD-NMR, computation, biochemical assays and click-chemistry, we have identified a chromogranin-A derived compound (5) that has high affinity and bi-selectivity for αvβ6 and αvβ8 integrins and is stable in microsomal preparations. 5 is suitable for nanoparticle functionalization and delivery to cancer cells, holding promise for diagnostic and/or therapeutic applications.
Integrins αvβ6 and αvβ8 are epithelial-specific cell-adhesion receptors, playing a fundamental role in pro-fibrotic cytokine transforming growth factor beta (TGFβ) activation in fibrosis.1 They are also highly expressed during tissue remodelling, wound healing, and cancer cell migration, invasion and growth, whereby over-expression correlates with poor patient prognosis.2,3 Hence, targeting of cells highly expressing one or both integrins through high affinity ligands with dual specificity and reduced off-targeting effects may represent a valid, yet poorly explored pharmacological strategy against cancer and/or fibrosis. αvβ6 and αvβ8 are structurally4 and functionally related,3 albeit αvβ85–7 and its inhibition is far less studied than αvβ6.8–13 Both integrins bind to arginine-glycine-aspartate (RGD) containing extracellular matrix proteins, whereby selective recognition occurs through the LXXL/I motif contiguous to the RGD sequence (RGDLXXL/I),5,14 which folds into one-helical turn upon binding to the receptor, thereafter engaging in specific lipophilic interactions with the hydrophobic pocket of the β6 or β8 subunit.5,15–18 We have previously shown that human chromogranin A (CgA), a neurosecretory protein involved in the cardiovascular system, metabolism, and tumor physiology19,20 regulation is a natural ligand of αvβ6.21 A CgA-derived peptide (residues 39–63) (1) also recognizes αvβ6 with nanomolar affinity and high selectivity (Ki: 15.5 ± 3.2 nM) (Table 1), herewith regulating αvβ6-dependent keratinocyte adhesion, proliferation, and migration.21 Notably, 1 harbours a degenerate RGDLXXL/I motif, with a glutamate replacing a leucine after the RGD sequence (position D + 1, RGDEXXL) (Fig. S1, ESI†). Prompted by this peculiarity, we investigated the structural determinants of 1/αvβ6 interaction by heteronuclear 2D-NMR STD methods and docking calculations. Intriguingly, while 1 is highly specific for αvβ6, reconstitution of the canonical RGDLXXL motif, combined with a click-chemistry stapling strategy results in a novel potent ligand suitable for the dual targeting of αvβ6/αvβ8 for diagnostic and therapeutic purposes.
| Code | Peptidea | αvβ6 | αvβ8 | α5β1 | αvβ5 | αvβ3 | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| n | K i | n | K i | n | K i | n | K i | n | K i | ||
| a Mutated residues and triazole-stapled residues (X1 and X2, as defined in Fig. S6a, ESI). b n, number of independent experiments (each performed with 6 different concentrations of competitor in technical duplicates). c K i of 2 as determined in ref. 21. d P value versus1: p < 0.05; two tailed t test. e P value versus1: p < 0.001, two tailed t test. f P value versus1: p < 0.01, two tailed t test. g P value versus4: p < 0.05; two tailed t test. h P value versus4: p > 0.1; two tailed t test. | |||||||||||
| 1 | FETLRGDERILSILRHQNLLKELQD | 6 | 15.5 ± 3.2 | 6 | 7663 ± 1704 | 4 | 9206 ± 1810 | 5 | 3600 ± 525 | 4 | 2192 ± 690 |
| 2 | FETLRGEERILSILRHQNLLKELQDc | 1 | >50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 000 |
1 | >50000 |
1 | >50000 |
1 | >50000 |
1 | >50000 |
| 3 | FETLRGDERILSILR | 4 | 277 ± 74d | 1 | 31174 |
1 | 10110 |
1 | 2039 | 1 | 1250 |
| 4 | FETLRGDLRILSILRHQNLLKELQD | 11 | 1.6 ± 0.3e | 6 | 8.5 ± 3.7f | 3 | 924 ± 198 | 4 | 2405 ± 592 | 3 | 1928 ± 226 |
| 5 | FETLRGDLRILSILRX1QNLX2KELQD | 7 | 0.6 ± 0.1g | 6 | 3.2 ± 1.2h | 3 | 1310 ± 389 | 4 | 2741 ± 615 | 3 | 2453 ± 426 |
| 6 | NAVPNLRGDLQVLAQKVART | 8 | 0.9 ± 0.2 | 6 | 69 ± 12 | 5 | 2317 ± 10 | 5 | 15449 ± 2418 |
3 | 26197 ± 7387 |
We studied the conformation of recombinant peptide 1 in physiological conditions by homonuclear and heteronuclear multidimensional NMR. Peptide 1 was expressed in E. coli as an insoluble fusion partner of ketosteroid isomerase, and subsequently cleaved with CNBr and purified by HPLC.22 Recombinant 13C/15N 1 displays the typical NOE pattern of the α-helical conformation between residue E46 to K59, with both termini being unstructured (Fig. 1a, c and Tables S1, S2, ESI†). Accordingly, the helical segment and both termini display relatively high (∼0.5) and very low (<0.3) heteronuclear NOE values, respectively (Fig. 1d). The RGD motif adjacent to the α-helix is relatively flexible, and thus well suited to adapt inside the integrin-binding pocket (Fig. 1a). The first three turns of the post-RGD helix are amphipathic, with I48, L49, I51, L52 and E46, R47, S50 on opposite sides (Fig. 1a and b). Peptide 1 propensity to adopt an α-helical conformation is in line with previous NMR studies on CgA47–66, an antifungal CgA-derived peptide, all-helical in the helix-promoting solvent tri-fluoro-ethanol, TFE.23 To gain molecular insights into the 1/αvβ6 complex and group selective information on the interaction, we performed in the presence of the extracellular region of recombinant human αvβ6 (4 μM) 1D-1H saturation transfer difference (STD) spectroscopy (Fig. S2a, ESI†) and heteronuclear two-dimensional STD experiments,24 exploiting isotopically labelled (13C/15N) recombinant peptide 1 (0.5 mM) (Fig. 2a and Fig. S2b, ESI†). The 2D-STD-1H–15N-HSQC resolved peak ambiguities in the 1D-1H STD spectrum and provided residue-specific STD effect values. Hydrophobic amino acids (I48, L49, I51, and L52) of the post-RGD helix displayed the strongest STD% values24 (>75%), suggesting their important contribution to receptor binding (Fig. 2b). Intense STD effects of the methyl groups of branched amino acids in 2D-STD-1H–13C-HSQC corroborated their involvement in the interaction, though signal overlap hampered their quantification for epitope mapping (Fig. S2b, ESI†). To exclude false positive effects, we spiked recombinant peptide 1 with bovine serum albumin as a negative control: no interaction occurred, and we did not observe any STD signal (Fig. S3a and b, ESI†). 2D-STD-1H–15N-HSQC performed with αvβ6 pre-incubated with 20 mM of EDTA resulted in depletion of the STD effects, thus confirming the presence of the electrostatic clamp between the receptor metal ion and the aspartate side chain of the RGD motif (Fig. S3c, ESI†). This result is in line with competitive binding assays using a peptide with RGE instead of RGD (2), yielding a Ki >50 μM (Table 1). Next, we incorporated the 2D-STD experimental information in data driven docking calculations (HADDOCK2.2)25 to determine the binding mode of 1 with the extracellular head of αvβ6 (PDB: 5FFO).16 The model highlights receptor–ligand interactions highly reminiscent of those observed for the proTGF-β1/αvβ6 complex (Fig. 2c and Fig. S4a, ESI†).16 On one hand, the guanidinium of R43 forms electrostatic interactions with Asp218αv and Asp150αv; on the other hand, the carboxylate of D45 coordinates the metal ion-dependent adhesion site (MIDAS) and interacts with the amide of Ser127β6 and Asn218β6. I48, L49, I51, and L52, located respectively on the second and the third turn of the post-RGD amphipathic α-helix, make extensive hydrophobic interactions with β6 residues of the specificity determining loops (SDLs), including Ala126β6, Asp129β6 (SDL1), Ile183β6, Tyr185β6 (SDL2), and Ala217β6 (SDL3) (Fig. 2c), thus explaining the selectivity of 1 towards αvβ6 with respect to the other αv integrins (Table 1). Since in our model residue E46 points towards the receptor interior, we reasoned that the preformed α-helix of 1 might entropically compensate the unfavourable electrostatic contribution of the negative charge within the hydrophobic binding pocket. Thus, we synthesized a shorter peptide containing the hydrophobic residues important for the interaction, without ten C-terminal residues supposed to be crucial for the helical propensity (3). Indeed, 3 showed a drastic reduction both in α-helical content (Fig. S5, ESI†) and binding to αvβ6 (Ki: 277 ± 77 nM) (Table 1), supporting the notion that the stability of the preformed four-turn amphipathic helix adjacent to the RGD motif is fundamental for effective αvβ6 recognition.18,26 We next predicted that restoring the canonical LXXL motif might increase the affinity of 1 for αvβ6. Indeed, the replacement in position D + 1 of E46 with a leucine (4) lowered the Ki by one order of magnitude (Ki: 1.6 ± 0.3 nM) (Table 1). Intriguingly, reconstitution of the LXXL motif transforms 4 into a bi-selective ligand able to bind also αvβ8 (Ki: 8.5 ± 3.7 nM).
 | ||
| Fig. 1 Solution structure of 1. (a) Representation of the 15 lowest energy NMR structures (pdb code: 6R2X) aligned on E46–N56 backbone atoms with the RGD motif in orange and I48, L49, I51 and L52 in green. (b) Helical wheel projection of residue E46–L52 with hydrophobic residues in green. (c) Scheme of medium and short NOEs relevant for secondary structure identification. Height of the boxes is proportional to NOE intensities. (d) Sequence specific backbone {1H}–15N NOEs with elements of secondary structure indicated on the top. | ||
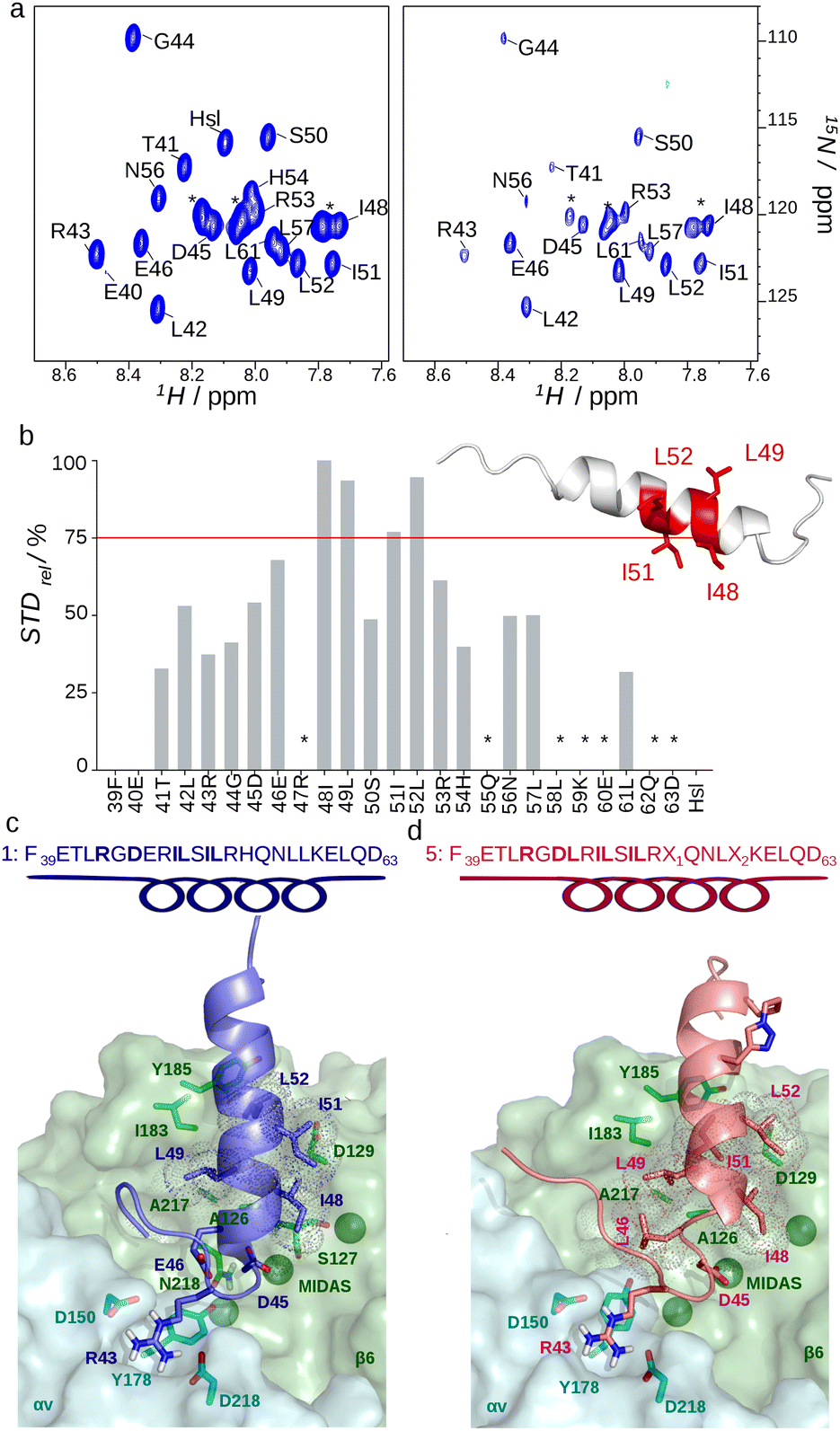 | ||
| Fig. 2 Interaction of αvβ6 with 1 and 5. (a) 2D-STD-1H–15N-HSQC experiment performed on 15N labelled peptide 1 (0.5 mM) in the presence of recombinant extracellular αvβ6 (4 μM), off-resonance (left) and difference spectra (right); asterisks indicate overlapped signals; Hsl (Homoserine lactone). (b) Residue-specific STD%, as defined in the ESI;† asterisks indicate overlapping signals. Residues with STD% > 75% are mapped on the 3D structure. (c) HADDOCK model of 1 (blue cartoon) and (d) 5 (pink cartoon) in complex with αvβ6. Ligand and receptor residues involved in the interaction, E46 in 1 and the triazole-containing stapled residues in 5 are shown in sticks. Sequence and secondary structure of 1 and 5 are shown on the top. Interacting residues are highlighted in bold. | ||
Structurally, αvβ6 and αvβ8 share a similar wide lipophilic SDL pocket, suitable for hydrophobic interactions with the amphiphilic helix of 4. Of note, minor changes in the shape and in the sequence of the SDL loops, such as the presence in SDL2 of K170 and T171 in β6 and S159 and I160 in β8, respectively (Fig. S4c, ESI†), might explain why the presence of E46 in the ligand is tolerated by β6 and not by β8 (Table 1). Prompted by these results, we hypothesized that chemical stabilization of the α-helix via stapling, i.e. “side-chain-to-side-chain” cyclization,27 might further improve the binding properties of 4. Based on a 5/αvβ6 model (Fig. 2d) we constrained this peptide via a triazole-bridged macrocyclic scaffold between residues in position 54 (propargylglycine) and 58 (azidolysine) through copper-catalyzed azide–alkyne cycloaddition (5) (Fig. S6a and b, ESI†).27,28 Indeed, the structural constraint boosted the α-helical content of 5, compared to 4, (Fig. S6c, ESI†), resulting in a significant 2 to 3-fold increase in αvβ6 binding (Ki: 0.6 ± 0.1 nM), comparable to the reference compound foot and mouth disease virus-derived peptide A20FMDV2 (6, Ki: 0.9 ± 0.2 nM) (Table 1).11,18 Stapling maintained nM binding to αvβ8 (Ki: 3.2 ± 1.2 nM), thus generating, to the best of our knowledge, the strongest bi-selective ligand for αvβ6/αvβ8 described so far.6,29 Importantly, peptides 1, 4, 5 and 6 were able to recognize αvβ6 in its physiological context, as they bound αvβ6 expressed on the cell-surface of human bladder 5637 cells and human keratinocytes (HaCat) with a relative binding potency similar to that observed with the purified recombinant αvβ6 (Fig. S7, ESI†). 5 was the most effective with an activity comparable to the reference compound 6 (Fig. 3a and Fig. S8, ESI†).30 Notably, both 4 and 5 were not cytotoxic in vitro (Fig. S9, ESI†). To assess whether 5 was suitable for nanoparticle functionalization and delivery to cancer cells, we coupled it to fluorescent quantum dot nanoparticles via an N-terminal cysteine (5-Qdot) and evaluated its binding to 5637 cells. Flow cytometry and fluorescence microscopy showed that 5-Qdot, but not a control nanoconjugate without the targeting ligand (*Qdot), bound the cells, indicating that 5 maintains its receptor-tailored properties also after conjugation (Fig. 3b and c). Finally, ELISA stability assays of 4 and 5 conjugated to horseradish peroxidase (4-HRP, 5-HRP) in human serum showed that >50% of 4-HRP and 5-HRP were still present after 24 hours of incubation at 37 °C, supporting their proteolytic stability in biological fluids (Fig. S10, ESI†). Importantly, stability assays with mouse liver microsomes showed that 5 was more stable than 4 (t1/2 = 4.3 h and t1/2 = 1.3 h, respectively, Fig. S11, ESI†).
 | ||
| Fig. 3 Binding of CgA-derived peptides to human bladder cancer 5637 cells. (a) Effect of 1, 4, 5 and 6 on the binding of anti-αvβ6 mAb 10D5 to 5637 cells. Antibody binding quantification by flow cytometry analysis (FACS) (representative experiment, see also Fig. S8a, ESI†). Compounds were mixed with mAb 10D5 and added to cells; mAb binding was detected by FACS and inhibitory concentration (IC50, mean ± SEM) was determined. Each point is in duplicate. (b) Binding of 5-Qdot or *Qdot (control) to 5637 cells as measured by FACS. Representative FACS (left and middle) and quantification of Qdot binding (right) (circles: mean ± SD of duplicates). (c) Representative fluorescence bioimaging of 5637 cells incubated with 5-Qdot, *Qdot or diluent. Magnification 40×; red, Qdot; blue, nuclear staining with DAPI. | ||
In conclusion, NMR experiments allied to computational and biochemical methods elucidated the molecular details at the basis of αvβ6 recognition by CgA-derived peptides, giving first hints on the interaction between αvβ6 and CgA.19 The entropic gain, derived from the preformed four-turns α-helix adjacent to the RGD motif, combined to the hydrophobic interactions between residues in position D + 3, D + 4, and D + 7 and the β6 subunit, largely compensate the unfavourable electrostatic repulsion of E46 in position D + 1. Thus, the natural αvβ6 recognition motif RGDLXXL is less restrictive than previously supposed and can be extended to RGDEXXL, provided that the helix adjacent to RGD is preformed and presents an extensive hydrophobic surface for αvβ6 interaction. Importantly, the complex model inspired the design of novel peptides, including a stapled one with high stability, sub-nanomolar affinity and bi-selectivity for αvβ6/αvβ8 integrins. These molecules, derived from a human protein, may represent useful and safer tools for the ligand-directed targeted delivery of diagnostic and therapeutic compounds and nanodevices to epithelial cancers with high expression of αvβ6 and/or αvβ8, such as oral and skin squamous cell carcinoma.31 Furthermore, in light of the roles of both αvβ6 and αvβ8 in TGFβ maturation and fibrosis,1 the dual targeting ability of these compounds could be also conveniently used to develop anti-fibrotic drugs and tracer devices, thus adding to the still limited number of small molecules able to specifically recognize these integrins.
The authors thank M. Alfano for helpful discussion. This work was supported by EU Horizon 2020 (801126, EDIT) and AIRC (IG-19220, IG-21440, 22737). F. N. conducted this study within her PhD course at S Raffaele University, Milan.
Conflicts of interest
There are no conflicts to declare.Notes and references
- K. P. Conroy, L. J. Kitto and N. C. Henderson, Cell Tissue Res., 2016, 365, 511–519 CrossRef CAS PubMed.
- L. Koivisto, J. Bi, L. Häkkinen and H. Larjava, Int. J. Biochem. Cell Biol., 2018, 99, 186–196 CrossRef CAS PubMed.
- M. Nieberler, U. Reuning, F. Reichart, J. Notni, H. Wester, M. Schwaiger, M. Weinmüller, A. Räder, K. Steiger and H. Kessler, Cancers, 2017, 9, 116 CrossRef PubMed.
- I. D. Campbell and M. J. Humphries, Cold Spring Harb. Perspect. Biol., 2011, 3, 1–14 Search PubMed.
- A. Ozawa, Y. Sato, T. Imabayashi, T. Uemura, J. Takagi and K. Sekiguchi, J. Biol. Chem., 2016, 291, 11551–11565 CrossRef CAS.
- F. Reichart, O. V. Maltsev, T. G. Kapp, A. F. B. Räder, M. Weinmüller, U. K. Marelli, J. Notni, A. Wurzer, R. Beck, H.-J. Wester, K. Steiger, S. Di Maro, F. S. Di Leva, L. Marinelli, M. Nieberler, U. Reuning, M. Schwaiger and H. Kessler, J. Med. Chem., 2019, 62, 2024–2037 CrossRef CAS PubMed.
- A. Cormier, M. G. Campbell, S. Ito, S. Wu, J. Lou, J. Marks, J. L. Baron, S. L. Nishimura and Y. Cheng, Nat. Struct. Mol. Biol., 2018, 25, 698–704 CrossRef CAS PubMed.
- O. V. Maltsev, U. K. Marelli, T. G. Kapp, F. S. Di Leva, S. Di Maro, M. Nieberler, U. Reuning, M. Schwaiger, E. Novellino, L. Marinelli and H. Kessler, Angew. Chem., Int. Ed., 2016, 55, 1535–1539 CrossRef CAS PubMed.
- F. S. Di Leva, S. Tomassi, S. DiMaro, F. Reichart, J. Notni, A. Dangi, U. K. Marelli, D. Brancaccio, F. Merlino, H. J. Wester, E. Novellino, H. Kessler and L. Marinelli, Angew. Chem., Int. Ed., 2018, 44, 14645–14649 CrossRef.
- M. Civera, D. Arosio, F. Bonato, L. Manzoni, L. Pignataro, S. Zanella, C. Gennari, U. Piarulli and L. Belvisi, Cancers, 2017, 9, 128 CrossRef PubMed.
- S. H. Hausner, D. DiCara, J. Marik, J. F. Marshall and J. L. Sutcliffe, Cancer Res., 2007, 67, 7833–7840 CrossRef CAS PubMed.
- R. H. Kimura, R. Teed, B. J. Hackel, M. A. Pysz, C. Z. Chuang, A. Sathirachinda, J. K. Willmann and S. S. Gambhir, Clin. Cancer Res., 2012, 18, 839–849 CrossRef CAS PubMed.
- A. Altmann, M. Sauter, S. Roesch, W. Mier, R. Warta, J. Debus, G. Dyckhoff, C. Herold-Mende and U. Haberkorn, Clin. Cancer Res., 2017, 23, 4170–4180 CrossRef CAS.
- S. Kraft, B. Diefenbach, R. Mehta, A. Jonczyk, G. A. Luckenbach and S. L. Goodman, J. Biol. Chem., 1999, 274, 1979–1985 CrossRef CAS.
- X. Dong, N. E. Hudson, C. Lu and T. A. Springer, Nat. Struct. Mol. Biol., 2014, 21, 1091–1096 CrossRef CAS.
- X. Dong, B. Zhao, R. E. Iacob, J. Zhu, A. C. Koksal, C. Lu, J. R. Engen and T. A. Springer, Nature, 2017, 542, 55–59 CrossRef CAS PubMed.
- A. Kotecha, Q. Wang, X. Dong, S. L. Ilca, M. Ondiviela, R. Zihe, J. Seago, B. Charleston, E. E. Fry, N. G. A. Abrescia, T. A. Springer, J. T. Huiskonen and D. I. Stuart, Nat. Commun., 2017, 8, 15408–15416 CrossRef CAS.
- D. DiCara, C. Rapisarda, J. L. Sutcliffe, S. M. Violette, P. H. Weinreb, I. R. Hart, M. J. Howard and J. F. Marshall, J. Biol. Chem., 2007, 282, 9657–9665 CrossRef CAS.
- A. Corti, F. Marcucci and T. Bachetti, Pflugers Arch. Eur. J. Physiol., 2018, 470, 199–210 CrossRef CAS PubMed.
- K. B. Helle, M. H. Metz-Boutigue, M. C. Cerra and T. Angelone, Pflugers Arch. Eur. J. Physiol., 2018, 470, 143–154 CrossRef CAS PubMed.
- F. Curnis, A. M. Gasparri, R. Longhi, B. Colombo, S. D’Alessio, F. Pastorino, M. Ponzoni and A. Corti, Cell. Mol. Life Sci., 2012, 69, 2791–2803 CrossRef CAS PubMed.
- J. L. Wagstaff, M. J. Howard and R. A. Williamson, Mol. BioSyst., 2010, 6, 2380–2385 RSC.
- K. Lugardon, S. Chasserot-Golaz, A.-E. Kieffer, R. Maget-Dana, G. Nullans, B. Kieffer, D. Aunis and M.-H. Metz-Boutigue, J. Biol. Chem., 2001, 276, 35875–35882 CrossRef CAS PubMed.
- J. L. Wagstaff, S. Vallath, J. F. Marshall, R. A. Williamson and M. J. Howard, Chem. Commun., 2010, 46, 7533 RSC.
- C. Dominguez, R. Boelens and A. M. J. J. Bonvin, J. Am. Chem. Soc., 2003, 125, 1731–1737 CrossRef CAS PubMed.
- Y. K. S. Man, D. DiCara, N. Chan, S. Vessillier, S. J. Mather, M. L. Rowe, M. J. Howard, J. F. Marshall and A. Nissim, PLoS One, 2013, 8, e70452 CrossRef CAS PubMed.
- Y. S. Tan, D. P. Lane and C. S. Verma, Drug Discov. Today, 2016, 21, 1642–1653 CrossRef CAS PubMed.
- A. Gori, C.-I. A. Wang, P. J. Harvey, K. J. Rosengren, R. F. Bhola, M. L. Gelmi, R. Longhi, M. J. Christie, R. J. Lewis, P. F. Alewood and A. Brust, Angew. Chem., Int. Ed., 2015, 54, 1361–1364 CrossRef CAS PubMed.
- T. G. Kapp, F. Rechenmacher, S. Neubauer, O. V. Maltsev, E. A. Cavalcanti-Adam, R. Zarka, U. Reuning, J. Notni, H. J. Wester, C. Mas-Moruno, J. Spatz, B. Geiger and H. Kessler, Sci. Rep., 2017, 7, 1–12 CrossRef PubMed.
- X. Huang, J. Wu, S. Spong and D. Sheppard, J. Cell Sci., 1998, 111, 2189–2195 CAS.
- H. Ahmedah, L. Patterson, S. Shnyder and H. Sheldrake, Cancers, 2017, 9, 56 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9cc08518a |
| ‡ Current address: University of Pisa, Via Moruzzi 13, I-56124 Pisa, Italy. |
| This journal is © The Royal Society of Chemistry 2019 |