
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelenoamides modulate dipole–dipole interactions in hydrogen bonded supramolecular polymers of 1,3,5-substituted benzenes†
José Augusto
Berrocal‡
 a,
Mathijs F. J.
Mabesoone‡
a,
Miguel
García Iglesias
abc,
Alex
Huizinga
a,
E. W.
Meijer
*a and
Anja R. A.
Palmans
*a
a,
Mathijs F. J.
Mabesoone‡
a,
Miguel
García Iglesias
abc,
Alex
Huizinga
a,
E. W.
Meijer
*a and
Anja R. A.
Palmans
*a
aInstitute for Complex Molecular Systems, Eindhoven University of Technology, P. O Box 513, 5600 MB Eindhoven, The Netherlands. E-mail: a.palmans@tue.nl; e.w.meijer@tue.nl
bDepartment of Organic Chemistry, Universidad Autónoma de Madrid (UAM), Calle Francisco Tomás y Valiente, 7, 28049 Madrid, Spain
cIMDEA Nanociencia, c/Faraday 9, Cantoblanco, Spain
First published on 16th November 2019
Abstract
We report the synthesis and self-assembly behavior of a chiral C3-symmetrical benzene-tricarboselenoamide. The introduction of the selenoamide moiety enhances the dipolar character of the supramolecular interaction and confers a remarkable thermal stability to the supramolecular polymers obtained.
Supramolecular chemistry has maturated up to a point where parallelisms between sophisticated multi-step covalent syntheses and the construction of increasingly complex, artificial supramolecular systems become more and more apparent.1–4 Recent developments in living supramolecular polymerizations,5–8 controlled supramolecular copolymerizations9–12 and the design of unconventional thermal behaviour in supramolecular polymers13–15 resulted in unprecedented control in length and microstructure of the formed polymers. Like in proteins, one of the central interactions underlying the stability and structure of many supramolecular systems is the formation of hydrogen bonds between amides. This ubiquitous interaction comprises several contributions such as dispersion interactions, electrostatics, polarization and charge transfer.16 Recently, it has become evident that the replacement of oxygen in amide bonds by other chalcogens such as sulphur and selenium retains the ability of the resulting thio- and selenoamides to engage in hydrogen bonding interactions.17–20 In addition, it has been observed both theoretically and experimentally that NH⋯S or NH⋯Se hydrogen bonds can be as strong as those of NH⋯O, despite the lower electronegativity of S and Se.21,22 In proteins and peptides, the introduction of thioamide or selenoamide bonds has allowed to tune their structure, stability, and – in case of enzymes – activity.23–26 Interestingly, as the chalcogen atom in the amide in peptides changes from O to S to Se, the increasing polarizability was found to increasingly dominate the electronic properties of the amides. As a result, the amide structure becomes increasingly characterized by the resonance form with negative charge on S or Se upon going down the periodic table.27 While the replacement of oxygen with sulphur typically results in a larger effect, the consequence of the further S to Se substitution is more modest, yet still detectable. As charge separation and dipolar interactions have been linked to cooperative behaviour in supramolecular polymers,28 selenoamides may provide interesting functionality in these non-covalent structures.
In the past we have extensively investigated the self-assembly of C3-symmetrical N,N′,N′′′-trialkylbenzene-1,3,5-tricarboxamides (OBTAs) in apolar alkane solvents (achiral OBTA and chiral c-OBTA, Scheme 1) and more recently also of N,N′,N′′′-trialkylbenzene-1,3,5-trithioamides (S-BTAs) (achiral SBTA and c-SBTA, Scheme 1).29,30
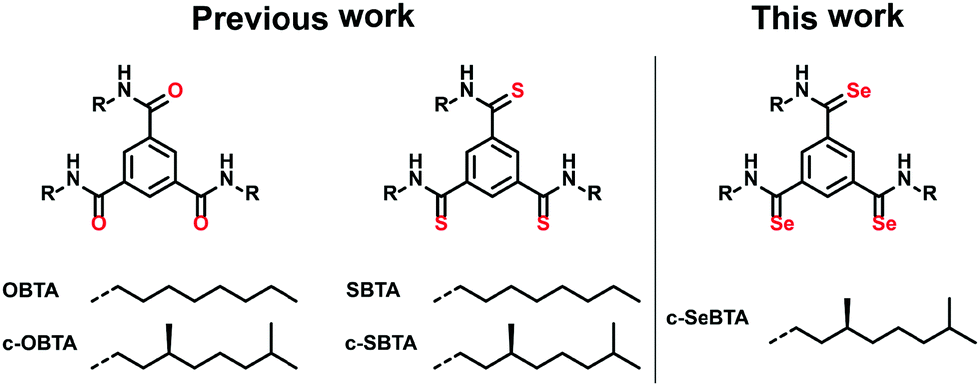 | ||
| Scheme 1 Chemical structures of previously reported OBTA, c-OBTA, SBTA and c-SBTA; chemical structure of the chiral selenoamide derivative c-SeBTA introduced in this work. Enantiopure compounds are indicated with the prefix c-. | ||
Stimulated by the increased dipolar character of the interactions between thioamides as compared to normal amides,31–33 we hypothesized that moving one row further down the periodic table to selenoamides would affect the stability of the formed cooperative supramolecular polymers. In addition, we anticipated that in copolymerizations the microstructure of the copolymers could be modulated.
Here, we disclose the synthesis (reported in the ESI†) and self-assembly of the chiral, C3-symmetrical benzene-tricarboselenoamide c-SeBTA (Scheme 1). We compare its self-assembly behavior to that of OBTA/c-OBTA20 and SBTA/c-SBTA.21 The different nature of the selenoamide hydrogen bond is illustrated by our finding that c-SeBTA polymers have an increased stability towards thermal denaturation yet enhanced lability towards polar solvents-induced denaturation.
The chiral tricarboselenoamide c-SeBTA was synthesized in 82% yield by refluxing c-OBTA with Woollins reagent (Ph2P2Se4) in toluene (Scheme S1, ESI†).34 We monitored the successful conversion to selenoamides by Fourier-transform infrared (FT-IR) spectroscopy. The disappearance of the parent carboxamide C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O vibration at 1634 cm−1 was coupled to the appearance of a new vibration centered at 1538 cm−1, indicative for the formation of the desired product (Fig. S3–S5, ESI†). The N–H stretching vibration also shifted from 3220 cm−1 to 3151 cm−1 in the c-OBTA to c-SeBTA conversion. Further details on the molecular characterization of c-SeBTA can be found in the ESI.†
O vibration at 1634 cm−1 was coupled to the appearance of a new vibration centered at 1538 cm−1, indicative for the formation of the desired product (Fig. S3–S5, ESI†). The N–H stretching vibration also shifted from 3220 cm−1 to 3151 cm−1 in the c-OBTA to c-SeBTA conversion. Further details on the molecular characterization of c-SeBTA can be found in the ESI.†
Chiral SeBTA was obtained as a yellow solid. The yellow color remained upon storing the compound under argon. However, exposing the samples to air for several hours resulted in an irreversible color change (Fig. S6, ESI†), suggesting decomposition. The low air stability of c-SeBTA was also coupled to poor thermal stability in ambient atmospheres, as we could not detect any transition upon heating or cooling due to decomposition of the samples by differential scanning calorimetry (DSC). Traces of c-OBTA detected in mass spectrometry suggest that c-SeBTA can react with moisture to form c-OBTA and other degradation products. Hence, the conversion of the tricarboxamides into triselenoamides implies particular attention concerning the compound's storage and preservation (under argon and preferably refrigerated).
We first studied the self-assembly behavior of c-SeBTA in apolar alkane solvents with ultraviolet-visible (UV-Vis) and circular dichroism (CD) spectroscopy. Upon dissolution of c-SeBTA in methylcyclohexane (MCH) at 50 μM and thorough degassing, an absorption maximum at 342 nm was observed in the UV-vis spectrum (Fig. 1a, bottom part). This maximum was red-shifted with respect to the spectrum of c-SeBTA in methanol (Fig. 1a, bottom part), suggesting self-assembly in the alkane solvent. In addition, a bisignate CD spectrum was obtained in MCH (maxima/minima at 357 nm, 297 nm and 252 nm), which corroborated the presence of aggregates characterized by a preferred helical sense (Fig. 1a, top part). AFM measurements confirmed that aggregates of c-SeBTA are one-dimensional supramolecular polymers (Fig. S7, ESI†).
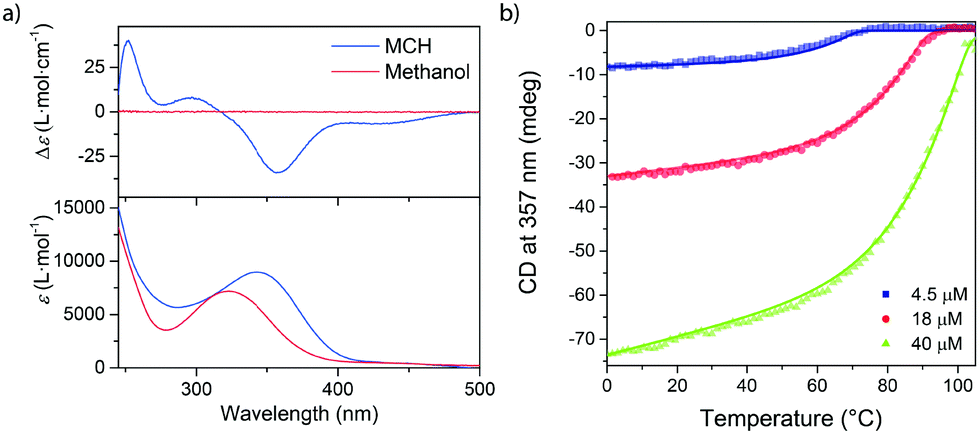 | ||
| Fig. 1 (a) CD (top) and UV-vis (bottom) spectra of 50 μM solutions of c-SeBTA in MCH and methanol (blue and red trace, respectively). (b) Variable temperature CD spectroscopy of solutions of c-SeBTA in MCH. Symbols indicate data and the solid lines indicate the global fit. | ||
We employed variable temperature CD (VT-CD) spectroscopy to investigate the stability of the supramolecular polymers of c-SeBTA. We followed the intensity of the CD band centered at 357 nm (Fig. 1b). Careful degassing of the prepared solutions was pivotal to ensure the reproducibility of the experiments (for the degassing procedure see ESI†). The 40 μM solutions of c-SeBTA showed CD activity up to temperatures as high as 100 °C, indicating the high thermal stability of the generated supramolecular polymers (Fig. 1b, green trace). Upon decreasing the concentration to 18 μM (Fig. 1b, red trace) and 4.5 μM (Fig. 1b, blue trace), a sharp transition between the CD-silent monomerically dissolved c-SeBTA to the supramolecular polymers of c-SeBTA was observed at 93 °C and 75 °C, respectively. This indicated a polymerization process characterized by a cooperative mechanism. By fitting the experimental data to a nucleation–elongation model,35 we determined enthalpy (ΔHe) and entropy (ΔSe) of elongation equal to −75 kJ mole−1 and −97 J mole−1 K−1, respectively, and a nucleation penalty of 21 kJ mole−1. The latter corresponds to a cooperativity factor, σ, of 2 × 10−4 at 293 K. Compared to the supramolecular polymers of c-SBTA (ΔHe = −65.7 kJ mole−1),30,36 the enthalpic contribution in the supramolecular polymerization of c-SeBTA was slightly more favorable, suggesting stronger intermolecular interactions in the case of the selenoamides. The entropic penalty of the supramolecular polymerization of c-SeBTA (ΔSe = −97 J mole−1 K−1) was comparable to that of c-SBTA30,36 (ΔSe = −102.6 J mole−1 K−1), instead, and we speculate similar ordering between the homopolymers of c-SeBTA and c-SBTA. We rationalize these thermodynamic parameters with an increased dipole–dipole and charge transfer character of the diffuse Se–NH hydrogen bond,21,37 which ultimately affords the increased thermal stability of the supramolecular polymers of c-SeBTA (more favorable ΔHe and very similar ΔSe compared to c-SBTA). Interestingly, the nucleation penalty, and hence cooperativity, of the supramolecular polymers of c-SBTA and c-SeBTA were comparable (σ = 1.9 × 10−3 for c-SBTA,30,36 and 3 × 10−4 for c-SeBTA). This is in strong contrast to the lower cooperativity of c–SBTA compared to c-OBTA (σ < 10−6).30,36
Next to VT-CD spectroscopy, the supramolecular polymerization of c-SeBTA was also investigated by changing the solvent quality in a denaturation experiment. Analogously to a previous study on c-SBTA38 discrete aliquots of CHCl3 (good solvent) were added to 20 μM solutions of c-SeBTA in MCH and the intensity of the CD signal was monitored (Fig. 2a and b). Upon increasing the solvent quality by adding CHCl3, the CD effect decreased in intensity (Fig. 2a). Above 4 vol%, no CD intensity could be observed anymore, indicating the complete destabilization of the polymers in 96![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 MCH:CHCl3 (Fig. 2a). When monitoring the maximum CD intensity at 357 nm, a clear non-sigmoidal dependency on the solvent quality is obtained (Fig. 2b), confirming the cooperative polymerization mechanism observed in the VT-CD experiments. The critical solvent fraction of 4 vol% CHCl3 in MCH at 20 μM for c-SeBTA is in remarkable contrast with a critical CHCl3 fraction of 20 vol% obtained for c-SBTA.38 This was unexpected, given the similar thermal behavior of the supramolecular polymers of c-SeBTA and c-SBTA discussed above. To obtain more insights into the thermodynamic properties of the denaturation process, the spectroscopic data were fitted to a mass-balance model that describes the destabilization of supramolecular polymers.38 For c-SeBTA, we obtain a Gibbs free energy of elongation (ΔGdene) of −36 kJ mole−1 in MCH, a solvent dependency parameter (m) of 602 kJ mole−1, as well as a nucleation penalty of 2.6 kJ mole−1. This Gibbs free energy of elongation is comparable to the Gibbs free energy of −40 kJ mole−1 reported for c-SBTA, but the solvent dependency parameter, m, obtained for c-SeBTA is six fold higher than the 103 kJ mole−1 reported for c-SBTA.38 We conclude that the enhanced charge separation in selenoamides27 increases the solubility in the relatively polar CHCl3 solvent. As a result, the interaction of the monomer with the polar co-solvent is enhanced and smaller amounts of co-solvent are needed to completely denature the polymer. Thus, on one side installing the selenoamide motif increases the thermal stability of the resulting supramolecular polymers in pure alkane solvents, while it simultaneously decreases their stability in more polar environments.
:4 MCH:CHCl3 (Fig. 2a). When monitoring the maximum CD intensity at 357 nm, a clear non-sigmoidal dependency on the solvent quality is obtained (Fig. 2b), confirming the cooperative polymerization mechanism observed in the VT-CD experiments. The critical solvent fraction of 4 vol% CHCl3 in MCH at 20 μM for c-SeBTA is in remarkable contrast with a critical CHCl3 fraction of 20 vol% obtained for c-SBTA.38 This was unexpected, given the similar thermal behavior of the supramolecular polymers of c-SeBTA and c-SBTA discussed above. To obtain more insights into the thermodynamic properties of the denaturation process, the spectroscopic data were fitted to a mass-balance model that describes the destabilization of supramolecular polymers.38 For c-SeBTA, we obtain a Gibbs free energy of elongation (ΔGdene) of −36 kJ mole−1 in MCH, a solvent dependency parameter (m) of 602 kJ mole−1, as well as a nucleation penalty of 2.6 kJ mole−1. This Gibbs free energy of elongation is comparable to the Gibbs free energy of −40 kJ mole−1 reported for c-SBTA, but the solvent dependency parameter, m, obtained for c-SeBTA is six fold higher than the 103 kJ mole−1 reported for c-SBTA.38 We conclude that the enhanced charge separation in selenoamides27 increases the solubility in the relatively polar CHCl3 solvent. As a result, the interaction of the monomer with the polar co-solvent is enhanced and smaller amounts of co-solvent are needed to completely denature the polymer. Thus, on one side installing the selenoamide motif increases the thermal stability of the resulting supramolecular polymers in pure alkane solvents, while it simultaneously decreases their stability in more polar environments.
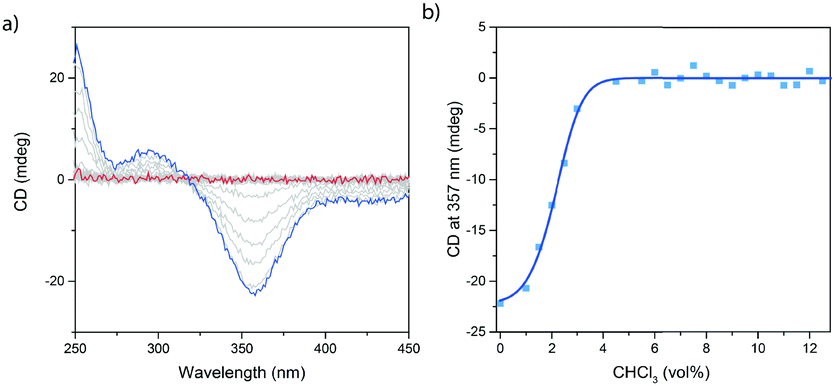 | ||
| Fig. 2 Denaturation experiment of c-SeBTA. (a) Change of the CD spectra of c-SeBTA (20 μM in MCH, blue line) upon adding CHCl3 (0–12 vol%, red line) and (b) CD effect at 357 nm as a function of the amount of CHCl3. The squares indicate experimental data and the solid lines represent the simulation. | ||
Besides influencing the stabilities of the supramolecular homopolymers, the different polarizability of oxygen, sulfur and selenium offers attractive possibilities to tune the interactions between monomers in supramolecular copolymers. We carried out mixed sergeants and soldiers experiment using c-SeBTA as sergeant for OBTA and SBTA to investigate the interaction between these supramolecular monomers (Fig. 3). The gradual titration of chiral c-SeBTA to a 30 μM solution of achiral OBTA in MCH resulted in the appearance of a CD spectrum typically observed for c-OBTA (Fig. S9a, ESI†), indicating that OBTA and c-SeBTA form copolymers. Complete chiral amplification was obtained by adding more than 30 mol% of c-SeBTA, as suggested by the saturation of the CD signal with respect to the intensity expected for a 30 μM solution of c-OBTA (Fig. 3 top panel). Analogously, addition of c-SeBTA to a 30 μM solution of SBTA resulted in the appearance of a CD spectrum typically observed for c-SBTA,36 which indicated successful copolymerization (Fig. S9b, ESI†). c-SeBTA was more effective in biasing the helical sense in the case of SBTA: only 12 mol% c-SeBTA was required to achieve full chiral amplification (Fig. 3 bottom panel). We applied the model developed by Markvoort and ten Eikelder10,39 to quantify the heterointeraction of c-SeBTA with OBTA and SBTA in the sergeants and soldiers experiment. These heterointeractions are dependent on the two homopolymerizations through an α-parameter (eqn (1)),30
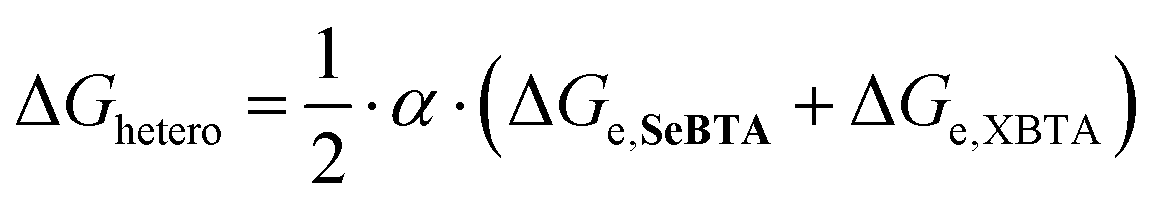 | (1) |
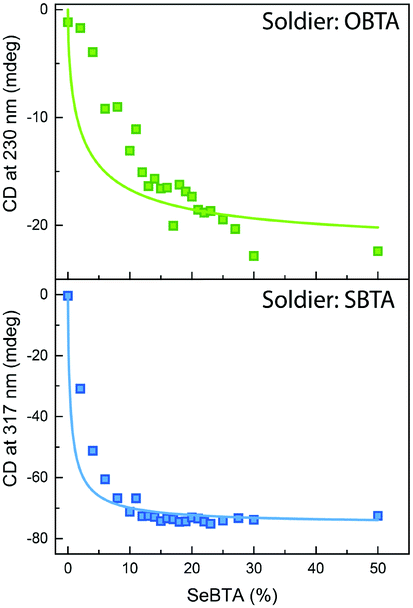 | ||
| Fig. 3 Sergeants and soldiers experiment of c-SeBTA sergeants with 30 mol% OBTA soldiers (top panel) and SBTA soldiers (bottom panel) in MCH. The squares indicate experimental data and the solid lines represent the simulation. | ||
The thermodynamic values for OBTA and SBTA have been described elsewhere.30 We obtained a good agreement between our experimental results and the model prediction for SBTA/c-SeBTA when using α = 0.815 (Fig. 3 bottom panel and Table 1). This value is lower than that previously reported for the nearly random copolymers of SBTA/OBTA (α = 0.92)29 but higher than those of the blocky copolymers formed by triarylamine-based systems (α = 0.57).10 This indicates that the copolymers of c-SeBTA/SBTA have a slightly blocky microstructure. In contrast, we could not find a good agreement between the model and the experimental data of OBTA/c-SeBTA (best fit for α = 0.72, Fig. 3 top panel). Most likely, the assumption made in the model that ΔGhetero is identical for OCNH⋯SeC and SeCNH⋯OC does not hold. The energetic difference between the XBTA homopolymerization (X = O or S) and the heterointeraction was smaller for the c-SeBTA/SBTA couple compared to the c-SeBTA/OBTA one (Table 1). The smaller difference in interaction strength for the neighboring chalcogens suggests that it is the polarizability of the groups that governs the interaction strength between the different monomers in the copolymerization. Hence, tuning heterointeractions by means of polarizability of the interacting co-monomers may prove to be an attractive way to tune supramolecular copolymerizations and control the microstructure of the resulting copolymers.
| OBTA | SBTA | |
|---|---|---|
| ΔGe,XBTA (kJ mole−1) | −33.6 | −36.4 |
| α | 0.72 | 0.815 |
| ΔGhetero (kJ mole−1) | −28.9 | −33.8 |
In conclusion, similarly to their oxygen and sulfur analogues, selenoamide-based discotic molecules self-assemble through hydrogen bond formation in apolar alkane solvents. The introduction of the selenoamides further increases the dipole–dipole and charge transfer interactions in the hydrogen bonding and continues a trend which was already observed upon changing carboxamides to thioamides.36 As a result, c-SeBTA shows an enhanced stability towards thermal denaturation in apolar alkane solvents while also showing an increased sensitivity towards polar co-solvents. Additionally, the subtle role played by polarizability and dipole–dipole interactions in hydrogen bonds offers an attractive way to design and control supramolecular copolymers. We showed successful copolymerization of c-SeBTA with OBTA and SBTA through sergeants and soldiers experiments. The interaction strength is highest for neighboring chalcogens, leading to the possibility to tune the supramolecular copolymerization through modulation of the dipole–dipole and charge transfer interactions in hydrogen bonds. We propose that our results will allow further control in the design of novel supramolecular polymers and serve as inspiration for further theoretical and experimental investigations into selenoamide hydrogen bonds.
Lafayette de Windt is gratefully acknowledged for providing the achiral OBTA and SBTA and for fruitful discussions. Tristan Mes and Elisa Huerta are acknowledged for the initial experiments. The authors thank NWO for financial support.
Conflicts of interest
There are no conflicts to declare.Notes and references
- N. Giuseppone, Acc. Chem. Res., 2012, 45, 2178–2188 CrossRef CAS PubMed.
- J. Lehn, Angew. Chem., Int. Ed., 2013, 52, 2836–2850 CrossRef CAS PubMed.
- P. Besenius, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 34–78 CrossRef CAS.
- G. Vantomme and E. W. Meijer, Science, 2019, 363, 1396–1397 CrossRef CAS PubMed.
- S. Ogi, K. Sugiyasu, S. Manna, S. Samitsu and M. Takeuchi, Nat. Chem., 2014, 6, 188–195 CrossRef CAS PubMed.
- T. Fukui, S. Kawai, S. Fujinuma, Y. Matsushita, T. Yasuda, T. Sakurai, S. Seki, M. Takeuchi and K. Sugiyasu, Nat. Chem., 2017, 9, 493–499 CrossRef CAS PubMed.
- W. Wagner, M. Wehner, V. Stepanenko, S. Ogi and F. Würthner, Angew. Chem., Int. Ed., 2017, 56, 16008–16012 CrossRef CAS PubMed.
- J. S. Valera, R. Gómez and L. Sánchez, Small, 2018, 14, 1702437 CrossRef PubMed.
- A. Pal, M. Malakoutikhah, G. Leonetti, M. Tezcan, M. Colomb-Delsuc, V. D. Nguyen, J. Van Der Gucht and S. Otto, Angew. Chem., Int. Ed., 2015, 54, 7852–7856 CrossRef CAS PubMed.
- B. Adelizzi, A. Aloi, A. J. Markvoort, H. M. M. Ten Eikelder, I. K. Voets, A. R. A. Palmans and E. W. Meijer, J. Am. Chem. Soc., 2018, 140, 7168–7175 CrossRef CAS PubMed.
- S. H. Jung, D. Bochicchio, G. M. Pavan, M. Takeuchi and K. Sugiyasu, J. Am. Chem. Soc., 2018, 140, 10570–10577 CrossRef CAS PubMed.
- S. Chakraborty, D. Ray, V. K. Aswal and S. Ghosh, Chem. – Eur. J., 2018, 24, 16379–16387 CrossRef CAS PubMed.
- T. Vermonden, J. Van Der Gucht, P. De Waard, A. T. M. Marcelis, N. A. M. Besseling, E. J. R. Sudhölter, G. J. Fleer and M. A. Cohen Stuart, Macromolecules, 2003, 36, 7035–7044 CrossRef CAS.
- K. Venkata Rao, D. Miyajima, A. Nihonyanagi and T. Aida, Nat. Chem., 2017, 9, 1133–1139 CrossRef CAS PubMed.
- V. Grande, B. Soberats, S. Herbst, V. Stepanenko and F. Würthner, Chem. Sci., 2018, 9, 6904–6911 RSC.
- T. Steiner, Angew. Chem., Int. Ed., 2002, 41, 48–76 CrossRef CAS.
- C. Alemán, J. Phys. Chem. A, 2001, 105, 6717–6723 CrossRef.
- H.-J. Lee, Y.-S. Choi, K.-B. Lee, J. Park and C.-J. Yoon, J. Phys. Chem. A, 2002, 106, 7010–7017 CrossRef CAS.
- D. Bibelayi, A. S. Lundemba, F. H. Allen, P. T. A. Galek, J. Pradon, A. M. Reilly, C. R. Groom and Z. G. Yav, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2016, 72, 317–325 CrossRef CAS PubMed.
- V. R. Mundlapati, S. Gautam, D. K. Sahoo, A. Ghosh and H. S. Biswal, J. Phys. Chem. Lett., 2017, 8, 4573–4579 CrossRef CAS PubMed.
- K. K. Mishra, S. K. Singh, P. Ghosh, D. Ghosh and A. Das, Phys. Chem. Chem. Phys., 2017, 19, 24179–24187 RSC.
- V. R. Mundlapati, D. K. Sahoo, S. Ghosh, U. K. Purame, S. Pandey, R. Acharya, N. Pal, P. Tiwari and H. S. Biswal, J. Phys. Chem. Lett., 2017, 8, 794–800 CrossRef CAS PubMed.
- T. T. Tran, J. Zeng, H. Treutlein and A. W. Burgess, J. Am. Chem. Soc., 2002, 124, 5222–5230 CrossRef CAS PubMed.
- D. Wildemann, C. Schiene-Fischer, T. Aumüller, A. Bachmann, T. Kiefhaber, C. Lücke and G. Fischer, J. Am. Chem. Soc., 2007, 129, 4910–4918 CrossRef CAS PubMed.
- Y. Huang, G. Jahreis, C. Lücke, D. Wildemann and G. Fischer, J. Am. Chem. Soc., 2010, 132, 7578–7579 CrossRef CAS PubMed.
- C. R. Walters, D. M. Szantai-Kis, Y. Zhang, Z. E. Reinert, W. S. Horne, D. M. Chenoweth and E. J. Petersson, Chem. Sci., 2017, 8, 2868–2877 RSC.
- Y. Huang, G. Jahreis, G. Fischer and C. Lücke, Chem. – Eur. J., 2012, 18, 9841–9848 CrossRef CAS PubMed.
- C. Kulkarni, S. Balasubramanian and S. J. George, ChemPhysChem, 2013, 14, 661–673 CrossRef CAS PubMed.
- M. M. J. Smulders, A. P. H. J. Schenning and E. W. Meijer, J. Am. Chem. Soc., 2008, 130, 606–611 CrossRef CAS PubMed.
- L. N. J. de Windt, C. Kulkarni, H. M. M. ten Eikelder, A. J. Markvoort, E. W. Meijer and A. R. A. Palmans, Macromolecules, 2019, 52, 7430–7438 CrossRef CAS PubMed.
- O. Exner and K. Waisser, Collect. Czech. Chem. Commun., 1982, 47, 828–837 CrossRef CAS.
- K. B. Wiberg and P. R. Rablen, J. Am. Chem. Soc., 1995, 117, 2201–2209 CrossRef CAS.
- K. B. Wiberg and D. J. Rush, J. Am. Chem. Soc., 2001, 123, 2038–2046 CrossRef CAS PubMed.
- P. Bhattacharyya and J. D. Woollins, Tetrahedron Lett., 2001, 42, 5949–5951 CrossRef CAS.
- D. Zhao and J. S. Moore, Org. Biomol. Chem., 2003, 1, 3471–3491 RSC.
- T. Mes, S. Cantekin, D. W. R. Balkenende, M. M. M. Frissen, M. A. J. Gillissen, B. F. M. De Waal, I. K. Voets, E. W. Meijer and A. R. A. Palmans, Chem. – Eur. J., 2013, 19, 8642–8649 CrossRef CAS PubMed.
- A. Kjaersgaard, J. R. Lane and H. G. Kjaergaard, J. Phys. Chem. A, 2019, 123, 8427–8434 CrossRef CAS PubMed.
- P. A. Korevaar, C. Schaefer, T. F. A. De Greef and E. W. Meijer, J. Am. Chem. Soc., 2012, 134, 13482–13491 CrossRef CAS PubMed.
- H. M. M. ten Eikelder, B. Adelizzi, A. R. A. Palmans and A. J. Markvoort, J. Phys. Chem. B, 2019, 123, 6627–6642 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details, synthesis and characterization of all new compounds, spectroscopic details, modeling details, temperature- and solvent-dependent supramolecular polymerization studies and fits. See DOI: 10.1039/c9cc08423a |
| ‡ These authors have contributed equally. |
| This journal is © The Royal Society of Chemistry 2019 |