
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Visible-light unmasking of heterocyclic quinone methide radicals from alkoxyamines†
Patrick
Kielty
 a,
Pau
Farràs
ab,
Patrick
McArdle
a,
Dennis A.
Smith
a and
Fawaz
Aldabbagh
*ac
a,
Pau
Farràs
ab,
Patrick
McArdle
a,
Dennis A.
Smith
a and
Fawaz
Aldabbagh
*ac
aSchool of Chemistry, National University of Ireland Galway, University Road, Galway, H91 TK33, Ireland
bEnergy Research Centre, Ryan Institute, National University of Ireland Galway, University Road, Galway, H91 CF50, Ireland
cDepartment of Pharmacy, School of Life Sciences, Pharmacy and Chemistry, Kingston University, Penrhyn Road, Kingston upon Thames, KT1 2EE, UK. E-mail: f.aldabbagh@kingston.ac.uk
First published on 14th November 2019
Abstract
In nature, the unmasking of heterocyclic quinones to form stabilized quinone methide radicals is achieved using reductases (bioreduction). Herein, an alternative controllable room-temperature, visible-light activated protocol using alkoxyamines and bis-alkoxyamines is provided. Selective synthetic modification of the bis-alkoxyamine, allowed chromophore deactivation to give one labile alkoxyamine moiety.
Nitroxides are bench-stable free radicals and anti-oxidants with a broad range of applications. The most widely used is commercial (2,2,6,6-tetramethylpiperidin-1-yl)oxyl (TEMPO) with applications in synthesis and catalysis,1a spin labelling,1b magnetic resonance imaging (MRI),1c fluorescence,1d electrochemistry,1e high-tech polymers,1f and radical scavenging.1g Nitroxides mask reactive carbon-centered radicals in the form of alkoxyamines. Homolysis of alkoxyamines is traditionally achieved via thermal activation, which for TEMPO usually requires temperatures above 80 °C (Scheme 1A).2 The high temperature reversible trapping of initiator-derived alkyl and polymer radicals by nitroxide, is a process made popular by controlled/living nitroxide-mediated polymerization (NMP).3 There are examples of NMP using UV-light driven photodissociation of alkoxyamines.4 From a biomedical applications perspective, alkoxyamine activation at or near physiological temperature is essential. Low or ambient temperature activation using UV/visible-light has been reported using alkoxyamine derivatives of 4-hydroxy-TEMPO (4-OH-TEMPO),4c,5 including XEt-TEMPOX.5c Guillaneuf and co-workers coupled benzylic derivatives of naphthalene, benzophenone, coumarin, anthraquinone and pyrene with 4-OH-TEMPO to give a series of UV/visible-light sensitive alkoxyamines (including the silylated TEMPO derivative).5b For reported light-sensitive alkoxyamines,4c,5 the formation of thermodynamically stabilized benzylic radicals promotes the loss of TEMPO. Comparably, the generation of a quinone methide drives enzymatic bioreduction of mitomycin C (MMC) to allow aziridinyl ring-opening and elimination of the carbamate functionality to give reactive sites for cross-linking with DNA.6 Benzimidazolequinone anti-tumor alternatives to MMC have been designed as prodrugs to form quinone methide upon bioreduction,7 or with adjustment of pH.8 Herein, we introduce heterocyclic benzimidazolequinone-based alkoxyamines; the simplest is TEMPO-Vis, where visible-light activated homolysis is driven by the formation of a quinone methide-type radical (QM-rad), stabilized by resonance delocalization (Scheme 1B). Bis-alkoxyamines are described, and visible-light is used for the first time to release up to two equivalents of TEMPO per molecule, using Bis-TEMPO-Vis. Bis- or bifunctional alkoxyamine activation has only been previously achieved by thermal means (at 70–140 °C),3,9 which for non-TEMPO bis-alkoxyamines leads to nitroxide decomposition.9c
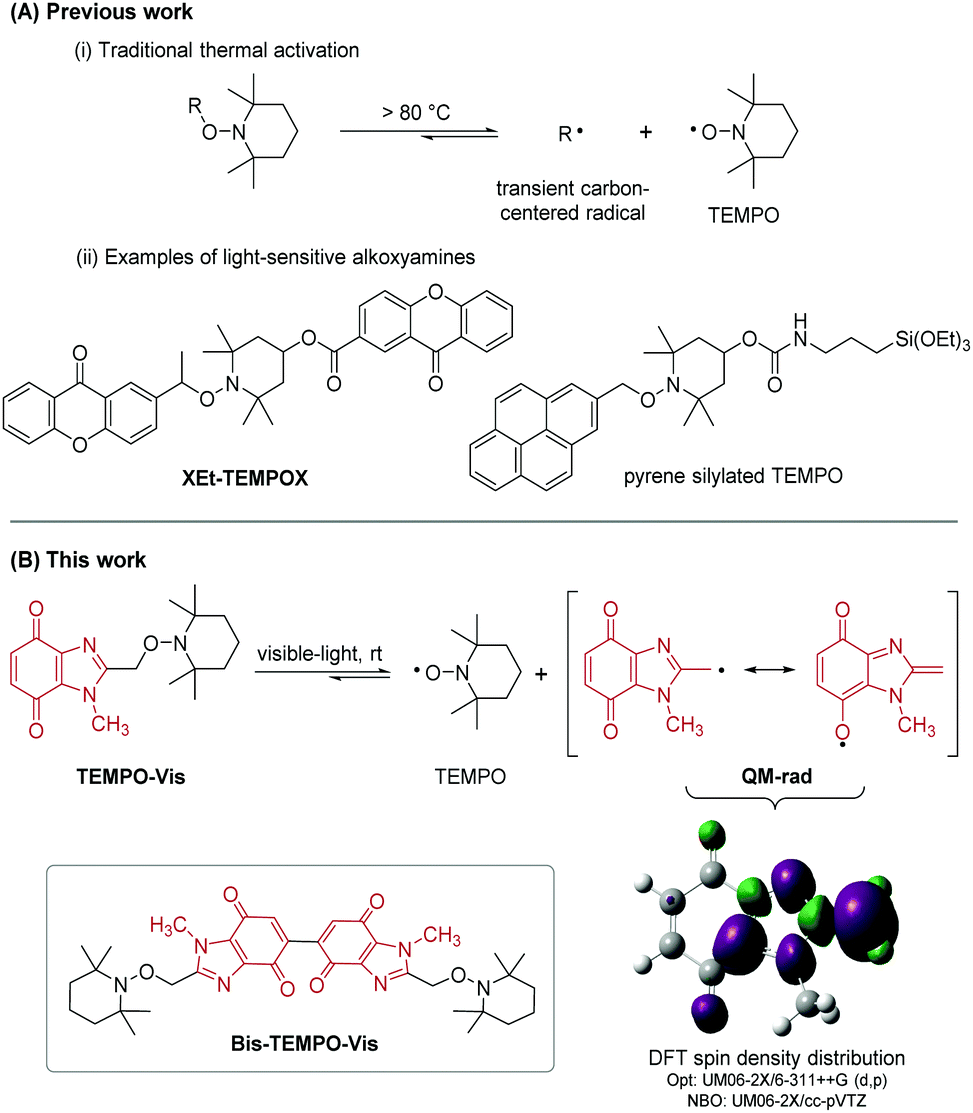 | ||
| Scheme 1 (A) Literature alkoxyamines (B) DFT-predicted controllable unmasking of heterocyclic quinone methide radicals using visible-light. | ||
The premise for this room temperature homolysis was obtained through DFT investigation of the level of delocalization of the unpaired electron in QM-rad (Fig. S1, ESI†). DFT supported the traditional resonance structures with a shortening of the 2C–CH2 bond relative to TEMPO-Vis, indicating partial double bond character (Table S1, ESI†), and significant distribution of spin density into the benzimidazolequinone ring system, including onto the quinone 7O-atom. The bond dissociation energy (BDE), and the lowest triplet energy level (ET) of TEMPO-Vis were estimated using DFT. The model-alkoxyamine (TEMPO-Vis) was found to have a suitably low BDE (85.1 kJ mol−1) compared to benzylic thermally labile (92.7–108.7 kJ mol−1),10 and UV-light activated (80–122 kJ mol−1)4a alkoxyamines, with enough driving force in the ET to support the observed bond dissociation: ΔGd = BDE − ET = −122.3 kJ mol−1 (see later discussion).
The preparation of TEMPO-Vis and Bis-TEMPO-Vis from 4,7-dimethoxybenzimidazole alkoxyamine precursor 1, using one-step oxidation was investigated (Table 1). Previous work used NBS in combination with H2SO4 to convert 4,7-dimethoxybenzimidazole into benzimidazolequinone.11 In the present work, TEMPO-Vis was isolated in 52% yield, with some undesirable bromination of 1 observed. PIFA [(CF3CO2)2IPh] improved the yield of TEMPO-Vis to 78%. Oxidative dimerization of dimethoxybenzenes to dibenzoquinones is reported through the use of CAN [Ce(NH4)2(NO3)6],12 and one report exists of a benzimidazolequinone dimer in low yield.13 Treatment of 1 with CAN (2 equiv.) afforded bis-alkoxyamine 2 in 86% yield, as the product of oxidative coupling of dimethoxybenzimidazole TEMPO-Vis with dimethoxybenz-imidazole 1. Increased amounts of CAN (3.2 equiv.) gave the fully oxidized Bis-TEMPO-Vis in 82% yield. The selective dimerization at C5–C5′ was confirmed by X-ray crystallography of Bis-TEMPO-Vis, with alternative couplings at C6–C6′, C5–C6′ and C6–C5′ not observed (Table 1).

To investigate the necessity in Bis-TEMPO-Vis of the benzimidazolequinone chromophore for the release of both attached TEMPO residues, a selective epoxidation strategy was devised for single chromophore deactivation (Scheme 2). Given its asymmetric aromatic/non-aromatic nature, 2 was deemed a good substrate for selective quinone functionalization. Subjecting 2 to the Langlois reaction (using NaSO2CF3),14 gave the electrophilic trifluoromethylated quinone bis-alkoxyamine 3 in 54% yield. The oxidative demethylation of 3 using NBS and H2SO4 (using Table 1 conditions), followed by mild epoxidation at the CF3-containing quinone moiety, via air oxidation under basic conditions, furnished epoxide-quinone 4 in 78% yield.
 | ||
| Scheme 2 Chromophore deactivation. | ||
Visible-light activated alkoxyamine homolysis was carried out under an O2 atmosphere to trap carbon-centered radicals,4a,5b,15 while monitoring alkoxyamine decay and TEMPO release by HPLC (Table 2).9b,16 Under blue LED (420–520 nm), 87% conversion of TEMPO-Vis to TEMPO was observed after 2.25 min. The decay of TEMPO-Vis alkoxyamine was first-order (Fig. 1A), from which the dissociation rate constant (kd) was determined, and corresponded to a half-life (t1/2) of 6 s. By monitoring [TEMPO] growth over time (Fig. 1B), eqn (1) may be fitted to the plot, to provide an alternative method to determine kd, which also gave a t1/2 of 6 s.
| [TEMPO] = [TEMPO]max(1 − e−kdt) | (1) |
| Alkoxyamine | LED color | k d via alkoxyamine decay (min−1) | k d via TEMPO release (min−1) | mol%d TEMPO released (time, min) | BDEe (kJ mol−1) | E T (kJ mol−1) |
|---|---|---|---|---|---|---|
| a Conditions: alkoxyamine (0.25 mM, DCE) illuminated at rt using blue (1 × 9 W) or green (2 × 9 W) LED bulbs under O2 balloon with HPLC analysis. Experiments performed in triplicate. b Dissociation rate (kd) derived from slope of Fig. 1A and Fig. S5A (ESI). c Derived from fit of eqn (1) to Fig. 1B and Fig. S5B (ESI). d HPLC yield based on starting alkoxyamine. e M06-2X, or UM06-2X for radicals, 6-311++G (d,p) in the gas phase. f BDE at benzimidazolequinone part. g BDE at epoxide/dimethoxybenzimidazole part. | ||||||
| TEMPO-Vis | Blue | 6.71 ± 0.21 | 7.29 ± 0.26 | 87 (2.25) | 85.1 | 207.4 |
| Bis-TEMPO-Vis | Blue | 2.78 ± 0.07 | 2.66 ± 0.09 | 175 (2.25) | 104.9 | 215.5 |
| 4 | Blue | 1.27 ± 0.16 | 1.41 ± 0.24 | 64 (2.25) | 99.7,f 104.6g | 209.7 |
| 2 | Blue | 0.00313 ± 0.00055 | 0.00417 ± 0.00024 | 78 (480) | 104.1,f 111.9g | 176.8 |
| TEMPO-Vis | Green | 0.0948 ± 0.0032 | 0.0993 ± 0.0063 | 80 (25) | — | — |
| Bis-TEMPO-Vis | Green | 0.148 ± 0.002 | 0.146 ± 0.004 | 162 (15) | — | — |
| 4 | Green | 0.0321 ± 0.0007 | 0.0346 ± 0.0061 | 43 (65) | — | — |
| 2 | Green | <2 × 10−4 | — | <5 (480) | — | — |
 | ||
Fig. 1 Kinetics (at rt) of alkoxyamine and bis-alkoxyamine homolysis in blue LED according to (A) (bis-)alkoxyamine decay and (B) TEMPO release. Eqn (1) was fitted to the plots in (B) using GraphPad Prism software. Key: TEMPO-Vis ( ), Bis-TEMPO-Vis ( ), Bis-TEMPO-Vis ( ) and 4 ( ) and 4 ( ). Conditions according to Table 2. ). Conditions according to Table 2. | ||
Alkoxyamines were indefinitely stable in the absence of light, and the on/off switchable nature of homolysis was demonstrated by alternating periods of light and dark for TEMPO-Vis using blue LED (Fig. S2, ESI†).
For the bis-alkoxyamine, Bis-TEMPO-Vis, there are two possible dissociation rate constants, kd1 and kd2 corresponding to TEMPO release from the starting compound, and from the monoalkoxyamine O2-trapped intermediate(s), with hydroperoxide and aldehyde intermediates detected by HPLC-MS (Fig. S3, ESI†). Just over twice as much TEMPO was released from Bis-TEMPO-Vis compared to TEMPO-Vis, with 175% released after the same period of 2.25 min. The kd1 was directly measured from the first-order decay plot of Bis-TEMPO-Vis, and corresponded to a t1/2 of 15 s in blue LED. The slower photolysis of Bis-TEMPO-Vis was supported by the DFT-derived ΔGd being 11.7 kJ mol−1 less favorable compared to TEMPO-Vis (Table 2). The rate of overall TEMPO release attained by fitting eqn (1) to the [TEMPO] vs. time plot (Fig. 1B), combines release from the starting bis-alkoxyamine (kd1), as well as from the O2-trapped intermediate alkoxyamine(s) (kd2). The rate constant derived in this way was in good agreement with the rate of Bis-TEMPO-Vis decay (Fig. 1A and Table 2). This infers that for Bis-TEMPO-Vis, kd1 ≈ kd2, and the homolysis of the alkoxyamine in the O2-trapped intermediate(s) occurs at an almost identical rate to that of the starting bis-alkoxyamine.
By removing one of the chromophores of Bis-TEMPO-Vis in epoxide-quinone 4, the release of <1 equiv. TEMPO occurred over the same time period (Table 2). The homolysis of one alkoxyamine of 4 was detected by HPLC-MS, with singly-homolyzed O2-trapped adducts observed (Fig. S3, ESI†), and no products of double alkoxyamine homolysis detected. Single bond homolysis occurred despite the BDE of the alkoxyamine of the epoxide part mirroring the BDE of Bis-TEMPO-Vis. TD-DFT calculations (see below) supported the localization of the frontier molecular orbitals on only the fully-conjugated quinone moiety of 4 (Fig. 2). The kd of the labile alkoxyamine of 4 is less than half that of Bis-TEMPO-Vis in blue LED, and given that the ΔGd values of the quinone-alkoxyamine in 4 and Bis-TEMPO-Vis are similar (at about −110 kJ mol−1), the observed reduction in rate may be attributed to the lower absorption of the partially deactivated 4 in the visible region (Fig. S4, ESI†).
 | ||
| Fig. 2 TD-DFT analysis of ground and excited state orbital delocalization in epoxide-quinone 4, p-dimethoxybenzene-coupled quinone 2, and Bis-TEMPO-Vis. Conditions: PCM/M06-2X/6-311++G (d,p), using DCE as solvent and the natural transition orbital (NTO)17 method for visualization. | ||
The rate of homolysis decreased using green (470–600 nm) compared to blue LED by 71-, 19- and 40-fold for TEMPO-Vis, Bis-TEMPO-Vis, and 4 respectively (Fig. S5, ESI†), due to less absorption. The decrease in absorbance is reflected in reduced quantum yields (Φh) with the greater intensity green LED used (see Table S2, ESI†). Moreover, Bis-TEMPO-Vis underwent photolysis at a faster rate than TEMPO-Vis in green LED, in accordance with λmax of the former being red-shifted by 12 nm (Fig. S4, ESI†). The result in green LED, is a reversal in the magnitude of kd that was observed for blue LED for the two compounds. The same conclusion of kd1 ≈ kd2 for Bis-TEMPO-Vis was reached for green LED activation.
Bis-alkoxyamine 2 was found to be largely stable under visible-light, having a kd three orders of magnitude smaller than its fully-oxidized derivative, Bis-TEMPO-Vis, in blue and green LED (Table 2). Although 2 possesses a similar first BDE to Bis-TEMPO-Vis, its ET was more than 30 kJ mol−1 lower than the other alkoxyamines. The λmax of 2 was red-shifted by 96 nm compared to Bis-TEMPO-Vis, suggesting the presence of a low-lying charge-transfer (CT) state (Fig. S4, ESI†). Cyclic voltammetry on 2 and its constituent alkoxyamines 1 and TEMPO-Vis, supported the localization of the HOMO and LUMO to the dimethoxybenzimidazole and the benzimidazolequinone motifs respectively (Fig. S6, ESI†). Time-dependent density functional theory (TD-DFT)18 provided graphical representation of spatially-separated ground and excited state orbitals (Fig. 2). The ground state (S0) of 2 is primarily localized on the dimethoxybenzimidazole, while the density of the first excited state (S1) is entirely localized on the quinone, with limited overlap between the two states. In comparison, the CT effect is not observed in the analogous TD-DFT of Bis-TEMPO-Vis.
In conclusion, alkoxyamines of heterocyclic quinones are introduced with room temperature visible-light homolysis providing an alternative to nature's bioreductive activation of prodrugs, as a means of unmasking the transient quinone methide. This includes an alkoxyamine that can release up to two equivalents of nitroxide per molecule using visible-light activation, and that does so sequentially with kd1 ≈ kd2. Facile synthetic deactivation of one chromophore limited TEMPO release to <1 equiv. For blue LED, the rates of bond homolysis can largely be rationalized by thermodynamics, while for green LED variations in absorbance become more important. The placement of an electron-rich substituent on the electron-deficient quinone gives a charge-transfer state that stabilizes the quinone under visible-light. The benzimidazolequinone alkoxyamines offer the possibility of wide-ranging applications from visible-light activated anti-tumour cytotoxins to radical initiators for vinyl monomer photopolymerizations giving polymers end-functionalized with antibiotics.
We thank the Irish Research Council for awarding PK an Enterprise Partnership Postgraduate Scholarship. We are grateful to Peter Cannon (Avara Pharmaceutical Services Ltd., Shannon, Ireland) for acting as the Enterprise Mentor. PF acknowledges support from Royal Society Newton Alumni programme. We thank Sylvester Byrne and Benjamin A. Chalmers for laboratory assistance, and John O’Reilly for consultation on HPLC (all National University of Ireland Galway). Andrew Benniston (Newcastle University, UK) is gratefully acknowledged for discussions around the photophysical and computational studies.
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) H. A. Beejapur, Q. Zhang, K. Hu, L. Zhu, J. Wang and Z. Ye, ACS Catal., 2019, 9, 2777 CrossRef CAS; (b) M. M. Haugland, J. E. Lovett and E. A. Anderson, Chem. Soc. Rev., 2018, 47, 668 RSC; (c) J. Wahsner, E. M. Gale, A. Rodríguez-Rodríguez and P. Caravan, Chem. Rev., 2019, 119, 957 CrossRef CAS PubMed; (d) A. Kaur, J. L. Kolanowski and E. J. New, Angew. Chem., Int. Ed., 2016, 55, 1602 CrossRef CAS PubMed; (e) J. E. Nutting, M. Rafiee and S. S. Stahl, Chem. Rev., 2018, 118, 4834 CrossRef CAS PubMed; (f) K.-A. Hansen and J. P. Blinco, Polym. Chem., 2018, 9, 1479 RSC; (g) E. G. Bagryanskaya and S. R. A. Marque, Chem. Rev., 2014, 114, 5011 CrossRef CAS PubMed.
- T. Fukuda, T. Terauchi, A. Goto, K. Ohno, Y. Tsujii, T. Miyamoto, S. Kobatake and B. Yamada, Macromolecules, 1996, 29, 6393 CrossRef CAS.
- J. Nicolas, Y. Guillaneuf, C. Lefay, D. Bertin, D. Gigmes and B. Charleux, Prog. Polym. Sci., 2013, 38, 63 CrossRef CAS.
- (a) D.-L. Versace, Y. Guillaneuf, D. Bertin, J. P. Fouassier, J. Lalevée and D. Gigmes, Org. Biomol. Chem., 2011, 9, 2892 RSC; (b) Y. Guillaneuf, D. Bertin, D. Gigmes, D.-L. Versace, J. Lalevée and J.-P. Fouassier, Macromolecules, 2010, 43, 2204 CrossRef CAS; (c) S. Hu, J. H. Malpert, X. Yang and D. C. Neckers, Polymer, 2000, 41, 445 CrossRef CAS.
- (a) A. Goto, J. C. Scaiano and L. Maretti, Photochem. Photobiol. Sci., 2007, 6, 833 RSC; (b) M. Baron, J. C. Morris, S. Telitel, J.-L. Clément, J. Lalevée, F. Morlet-Savary, A. Spangenberg, J.-P. Malval, O. Soppera, D. Gigmes and Y. Guillaneuf, J. Am. Chem. Soc., 2018, 140, 3339 CrossRef CAS PubMed; (c) M. Herder and J.-M. Lehn, J. Am. Chem. Soc., 2018, 140, 7647 CrossRef CAS PubMed.
- P. D. Bass, D. A. Gubler, T. C. Judd and R. M. Williams, Chem. Rev., 2013, 113, 6816 CrossRef CAS PubMed.
- (a) W. G. Schulz, R. A. Nieman and E. B. Skibo, Proc. Natl. Acad. Sci. U. S. A., 1995, 92, 11854 CrossRef CAS PubMed; (b) C. Flader, J. Liu and R. F. Borch, J. Med. Chem., 2000, 43, 3157 CrossRef CAS PubMed.
- E. B. Skibo, J. Org. Chem., 1992, 57, 5874 CrossRef CAS.
- (a) I. Q. Li, B. A. Howell, R. A. Koster and D. B. Priddy, Macromolecules, 1996, 29, 8554 CrossRef CAS; (b) G. Ananchenko and K. Matyjaszewski, Macromolecules, 2002, 35, 8323 CrossRef CAS; (c) J. Ruehl, N. L. Hill, E. D. Walter, G. Millhauser and R. Braslau, Macromolecules, 2008, 41, 1972 CrossRef CAS PubMed.
- J. L. Hodgson, L. B. Roskop, M. S. Gordon, C. Y. Lin and M. L. Coote, J. Phys. Chem. A, 2010, 114, 10458 CrossRef CAS PubMed.
- L. O'Donovan, M. P. Carty and F. Aldabbagh, Chem. Commun., 2008, 5592 RSC.
- B. E. Love, J. Bonner-Stewart and L. A. Forrest, Synlett, 2009, 813 CrossRef CAS.
- A. Gellis, H. Kovacic, N. Boufatah and P. Vanelle, Eur. J. Med. Chem., 2008, 43, 1858 CrossRef CAS PubMed.
- B. R. Langlois, in Modern Synthesis Processes and Reactivity of Fluorinated Compounds, ed. H. Groult, F. R. Leroux and A. Tressaud, Elsevier, 2017, ch. 5, pp. 125 Search PubMed.
- D. W. Grattan, D. J. Carlsson, J. A. Howard and D. M. Wiles, Can. J. Chem., 1979, 57, 2834 CrossRef CAS.
- G. Moad and E. Rizzardo, Macromolecules, 1995, 28, 8722 CrossRef CAS.
- R. L. Martin, J. Chem. Phys., 2003, 118, 4775 CrossRef CAS.
- A. D. Laurent and D. Jacquemin, Int. J. Quantum Chem., 2013, 113, 2019 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Supporting figures and tables, experimental procedures and NMR spectra. CCDC 1948448. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9cc08261a |
| This journal is © The Royal Society of Chemistry 2019 |