
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA study on the effect of synthetic α-to-β3-amino acid mutations on the binding of phosphopeptides to 14-3-3 proteins†
Sebastian A.
Andrei
 a,
Vito
Thijssen
a,
Luc
Brunsveld
a,
Christian
Ottmann
ab and
Lech-Gustav
Milroy
a
a,
Vito
Thijssen
a,
Luc
Brunsveld
a,
Christian
Ottmann
ab and
Lech-Gustav
Milroy
a
aLaboratory of Chemical Biology, Department of Biomedical Engineering and Institute for Complex Molecular Systems (ICMS), Eindhoven University of Technology, 5600 MB Eindhoven, The Netherlands. E-mail: l.brunsveld@tue.nl; c.ottmann@tue.nl; l.milroy@cantab.net
bDepartment of Chemistry, University of Duisburg-Essen, 47057 Essen, Germany
First published on 15th November 2019
Abstract
Here we describe the synthesis of a series of α,β-phosphopeptides, based on the phosphoepitope site on YAP1 (yes-associated protein 1), and the biochemical, biophysical and structural characterization of their binding to 14-3-3 proteins. The impact of systematic mono- and di-substitution of α → β3 amino acid residues around the phosphoserine residue are discussed. Our results confirm the important role played by the +2 proline residue in the thermodynamics and structure of the phosphoepitope/14-3-3 interaction.
14-3-3 proteins (a highly homologous family of 7 isoforms with overlapping functionalities) are promising drug targets for treating cancer and neurodegeneration,1–4 firstly because of their role as hub proteins within a number of signalling pathways critical to pathologies. Secondly, 14-3-3 proteins are eminently druggable in the sense that they preferentially bind to client proteins via conserved phosphorylated epitopes,5,6 which form well-defined drug interfaces7,8 for studies into 14-3-3 protein–protein interaction (PPI) inhibition or stabilization.9–11 Such studies have laid foundations for the discovery of small molecule modulators of 14-3-3 PPIs through e.g. the discovery of natural product stabilizers,9,12 the development of peptidomimetics,13,14 modified peptides,10 or small molecule inhibitors.15,16 The peptide epitope of a target PPI serves as an excellent entry point for the structure-based design of novel PPI inhibitors,13 including for 14-3-3 PPIs.10 Whereas the amino acid sequence and thus sidechain diversity of the 14-3-3 binding phosphopeptide has been extensively studied,6,17 the peptide backbone of the phosphoepitope has received less attention despite the clear potential for improved affinity and selectivity between highly homologous PPIs.18 Backbone modifications can improve both the pharmacodynamic and –kinetic properties of peptide modulators.19 α,β-Peptides are a particularly promising form of backbone modification.18,20–23 To the best of our knowledge α,β-peptides have so far not been studied in the context of 14-3-3 proteins. Here we report a comprehensive and systematic biochemical and structural study on the effects of α → β3 mutations on the binding of the phosphopeptide epitope of the WW-domain protein YAP1 to 14-3-3 proteins.
The yes-associated protein homolog, YAP1, is important for transcription regulation and 14-3-3 regulates trafficking of YAP1 between the cytoplasm and nucleus through binding to phosphorylated Ser127 on the YAP1 protein.24 YAP1 exhibits tumour promoting properties, which are suppressed by phosphorylation of residue Ser127 through subsequent binding to 14-3-3 proteins.25 Small molecule modulators of 14-3-3/YAP1 would be useful tool compounds and may serve as potential lead compounds for drug development. The crystal structure of the isoform 14-3-3σ/YAP1 binding interface was solved by our group.24 The 14-3-3/YAP1 was considered to be an ideal PPI to investigate σ/β-phosphopeptides because of the long extended interface leading to multiple contact points between peptide and 14-3-3 protein allowing to gauge the significance of the corresponding α-amino acids or region of the PPI.
In the original 14-3-3/YAP1 crystal structure [PBD: 3MHR], the 10-mer phosphopeptide sequence, bearing a C-terminal carboxylate and a free N-terminus (Table 1, YAP1.1), was co-crystallized with 14-3-3, and all amino acids of the YAP1 peptide could be fitted into the available electron density. A short study using fluorescence polarization (FP) and isothermal calorimetry (ITC) on the binding of the YAP1 peptide to two different 14-3-3 isoforms – ζ & σ, ideally suited for biophysical and crystallization studies10,11 – with either N-acetylation (YAP1.2) or C-amidation (YAP2.1), or both simultaneously (YAP2.2) – concluded that such modifications produce only a marginal change in 14-3-3-binding activity compared to YAP1.1 (Table 1; FP-curves in ESI†). Subsequent mutational studies were performed on YAP2.2.
| FP IC50/μM (±) | PDB code: | |||
|---|---|---|---|---|
| 14-3-3ζ | 14-3-3σ | |||
| a 14-3-3 isoform (1 μM), competitor FITC-ERα peptide9 (0.1 μM), starting concentration of dilution series = (250 μM). b 14-3-3 isoform (5 μM), competitor FITC-ERα peptide (0.1 μM), starting concentration of dilution series = (500 μM). YAP2.1, YAP2.2, βYAP6, and βYAP7 are against the limits of the assay. | ||||
| α-Peptides: | ||||
| H2N-RAHpSSPASLQ-COOH | YAP1.1 | 3.7 (0.33)a | 4.4 (0.14)a | 3MHR |
| Ac-RAHpSSPASLQ-COOH | YAP1.2 | 1.7 (0.03)a | 2.9 (0.10)a | — |
| H2N-RAHpSSPASLQ-NH2 | YAP2.1 | 1.1 (0.05)a | 1.9 (0.13)a | — |
| Ac-RAHpSSPASLQ-NH2 | YAP2.2 | 0.84 (0.02)a | 0.90 (0.40)a | — |
| α/β-Peptides: | ||||
RAHpS![[S with combining low line]](https://www.rsc.org/images/entities/b_char_0053_0332.gif) PASLQ PASLQ |
βYAP1 | >500b | >500b | — |
RAHpSS![[P with combining low line]](https://www.rsc.org/images/entities/b_char_0050_0332.gif) ASLQ ASLQ |
βYAP2 | 60.6 (3.8)b | 49.9 (8.3)b | 6G6X |
RAHpSSP![[A with combining low line]](https://www.rsc.org/images/entities/b_char_0041_0332.gif) SLQ SLQ |
βYAP3 | 17.2 (0.4)b | 12.4 (0.5)b | 6G8J |
| RAHpSSPALQ |
βYAP4 | 35.4 (2.3)b | 29.3 (0.5)b | 6G8K |
RAHpSSPAS![[L with combining low line]](https://www.rsc.org/images/entities/b_char_004c_0332.gif) Q Q |
βYAP5 | 10.0 (0.5)b | 7.7 (0.4)b | 6G8L |
RAHpSSPASL![[Q with combining low line]](https://www.rsc.org/images/entities/b_char_0051_0332.gif) |
βYAP6 | 5.9 (0.2)b | 6.7 (0.2)b | — |
![[R with combining low line]](https://www.rsc.org/images/entities/b_char_0052_0332.gif) AHpSSPASLQ AHpSSPASLQ |
βYAP7 | 5.8 (0.2)b | 6.8 (0.4)b | 6G8I |
| RHpSSPASLQ |
βYAP8 | 12.8 (0.3)b | 14.9 (0.7)b | — |
| RAHpSSASQ |
βYAP2.2 | 61.2 (3.5)b | 53.8 (3.4)b | 6G8P |
| RAHpSSPSL |
βYAP3.2 | 19.0 (0.3)b | 16.6 (0.8)b | 6G8Q |
We systematically mutated α → β3 amino acid residues in YAP2.2, except for phosphoserine and histidine residues, due to the lack of a suitable commercially available source. Eight α/β-phosphopeptides bearing a single α → β mutation, and two analogues bearing two α → β mutations were synthesized by Fmoc SPPS and purified and characterized by RP-HPLC (Table S1, ESI†). The eight singly mutated α/β-phosphopeptides were first tested in a competitive fluorescence polarization assay and their activity compared against YAP2.2 (Table 1). The data revealed an increase in the IC50 value the closer α → β3-substitution was made to the phosphoserine residue. As can be observed for βYAP1, replacing the α-serine with a β3-serine residue adjacent to the phosphoserine completely disrupts binding to 14-3-3σ. An α → β3-substitution one position further either N- or C-terminal to the phosphoserine, i.e.βYAP8 (β3-alanine) and βYAP2 (β3-proline), was correspondingly better tolerated – N-terminal more so than C-terminal. This might perhaps be unsurprising given that the peptides bear more amino acids C-terminal to the phosphate and thus more amino acids are able to interact with the surface of the 14-3-3 protein C-terminal to the phosphoserine. The hypothesis then being that α → β3-substitution leads to greater flexibility in the peptide backbone and thus a higher potential of disrupting peptide-protein interactions in this region of the peptide. This same principle holds when comparing α/β-phosphopeptides βYAP3 (β3-alanine) and βYAP7 (β3-arginine), in which the N-terminal substitution is apparently also more tolerated. The only exception to the above trend lies with βYAP4, whose activity is less than βYAP3 despite bearing an α → β3-substitution one position further away from the phosphoserine. Peptides βYAP2.2 and βYAP3.2, bearing two α → β3 amino acid mutations – one close and one further away from the phosphoserine – appear to exhibit the same activity as the connected βYAP2 and βYAP3 peptides, respectively. α → β3-mutations in the flanking region of the C-terminus have little effect in both the mono-substituted (βYAP5 and βYAP6) and di-substituted analogues (βYAP2.2 and βYAP3.2).
ITC measurements were performed on YAP2.2 (ESI†) and representative α → β3-peptides βYAP2 and βYAP4 to understand the biophysical consequences of α → β3-substitutions for binding to two different 14-3-3 isoforms – 14-3-3σ (ESI†) and 14-3-3ζ (Fig. 1). The binding of βYAP2 to 14-3-3ζ (Kd = 13.30 ± 1.13 μM) was found to be approximately two-fold weaker compared to that of βYAP4 (Kd = 6.71 ± 0.43 μM). A similar pattern of behaviour was observed toward the 14-3-3σ isoform (ESI†). The activity of these two α/β-phosphopeptides is approximately 10-fold weaker than observed for the reference peptide YAP2.2 (Kd = 0.79 ± 0.09 μM). Interestingly, though, this loss of activity is the result of a significantly more positive entropic contribution in the case of βYAP2 (i.e. ΔH = −9.0 ± 0.2 kcal mol−1, −TΔS = 2.0 kcal mol−1) in contrast to a significantly less negative enthalpic contribution in the case of βYAP4 (i.e. ΔH = −7.6 ± 0.08 kcal mol-1, −TΔS = 0.2 kcal mol−1), relative to YAP2.2 (i.e. ΔH = −9.5 ± 0.08 kcal mol−1, −TΔS = 0.8 kcal mol−1). These data suggest that the activity drop observed on introduction of the β3-amino acid thus might equally be explained by either a disruption to energetically favourable binding interactions between α/β-phosphopeptide and 14-3-3 protein or an increase in the degrees of conformational freedom in the peptide backbone.
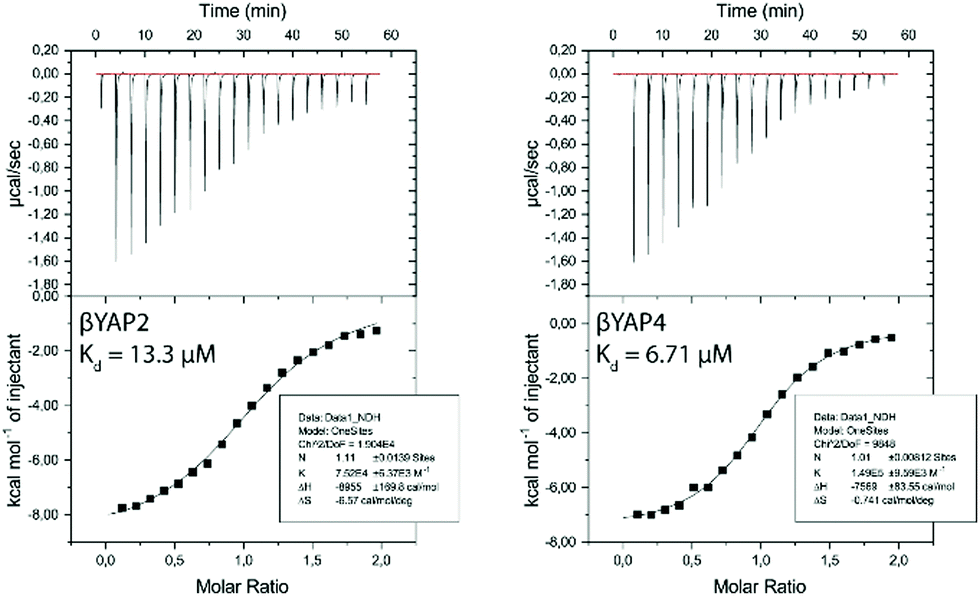 | ||
| Fig. 1 ITC measurements of the binding of α/β-phosphopeptides βYAP2 & βYAP4 to 14-3-3ζ (T = 310.15 K or 37 °C). Additional measurements of these ITC results can be found in the ESI.† | ||
A competitive fluorescence depolarization assay of all the α/β-phosphopeptides versusYAP2.2 was performed against five different 14-3-3 homodimer isoforms – σ,ζ,ε,η and τ – to investigate for a possible isoform selective behaviour of the peptides (Fig. S5–S9, ESI†). In general, the affinity of the α/β-phosphopeptides toward 14-3-3ε and 14-3-3η is greater than toward the other isoforms. The preference for 14-3-3η is present in all peptides including the α/β-peptides. Substitution of the proline (βYAP2), alanine (βYAP3) and serine residues (βYAP4) C-terminal to the phosphoserine residue showed a similar pattern of behaviour across the five isoforms (order of affinity, 14-3-3η > 14-3-3ε > 14-3-3σ >14-3-3ζ or 14-3-3τ). This affinity trend is also shared by the double mutant peptides (βYAP2.2 & βYAP3.2), though less pronounced, but is not observed for the α-peptide lacking β3-substitution i.e.YAP2.2.
To gain structural insight into the activity trends observed in the FP and ITC assays, the crystal structure for five of the eight α/β-phosphopeptides bearing a single β3-amino acid mutation – βYAP2, βYAP3, βYAP4, βYAP5, βYAP7 – and both peptides bearing two doubly mutated variants – i.e.βYAP2.2 & βYAP3.2 was solved (Fig. 2). A non-linear correlation was observed between the position of the α → β3 mutation and the amount of observable electron density C-terminal to the phosphorylation site. The amount of observed electron density decreased the further away from the phosphoserine residue a mutation was situated (βYAP2 → 4), only to increase again beyond βYAP5. In the case of βYAP2, bearing a β3-proline residue +2 to the phosphoserine, despite the mutation, we unexpectedly observe the whole peptide in the structure. On comparison of βYAP2 with native structure (YAP1.1, PDB 3MHR, Fig. 3) we observe that the introduction of the β3-proline residue results in a significant remodelling of the peptide conformation, particularly in the C-terminal region. Therefore, the Pro residue might be considered critical for determining the peptide's preferred bound conformation. For βYAP3 (β3-alanine) the introduction of the β3-amino acid residue, which is clearly visible in the electron density map, results in a loss of electron density in the C-terminal region beyond the mutated residue, suggesting one conformation for the peptide in the groove that is flexible beyond the mutated site. The introduction of a β3-serine residue by contrast, i.e.βYAP4, introduces considerable disorder at the peptide C-terminus, starting at the proline residue. The density of the peptide backbone is seen to branch into two conformations, both of which cannot be modelled beyond the proline branching point. For βYAP5 (β3-leucine) the whole peptide is visible in a very similar conformation to the conformation observed in the native sequence (YAP1.1).
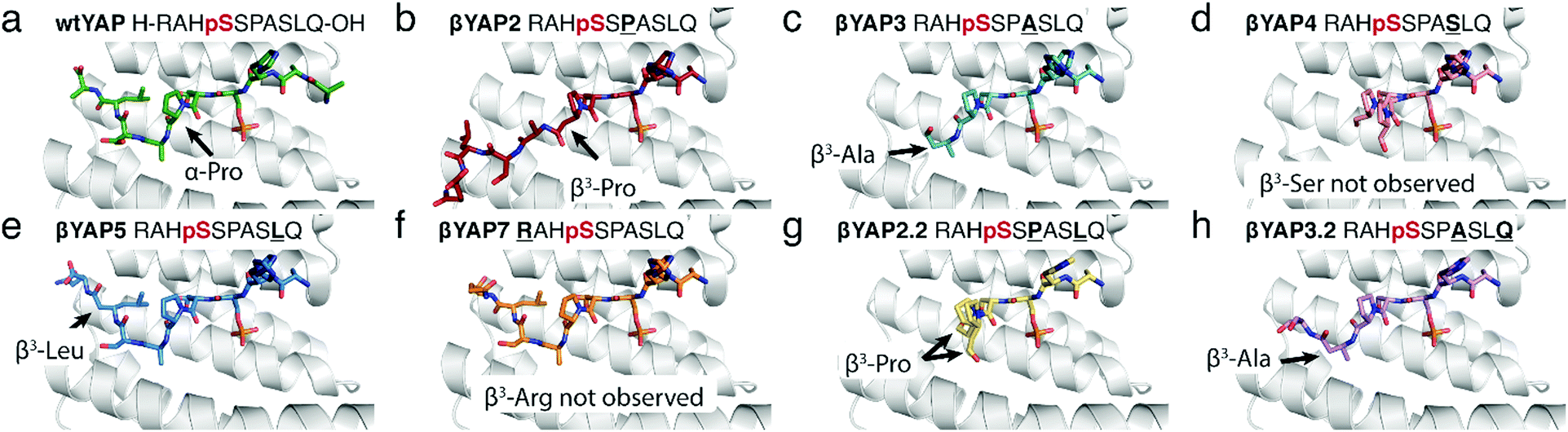 | ||
| Fig. 2 Overview of the crystal structures of 14-3-3σΔC (grey) in complex with (a) wildtype αYAP 10-mer peptide [PDB 3MHR] and (b–h) βYAP 10-mer peptides from this study. | ||
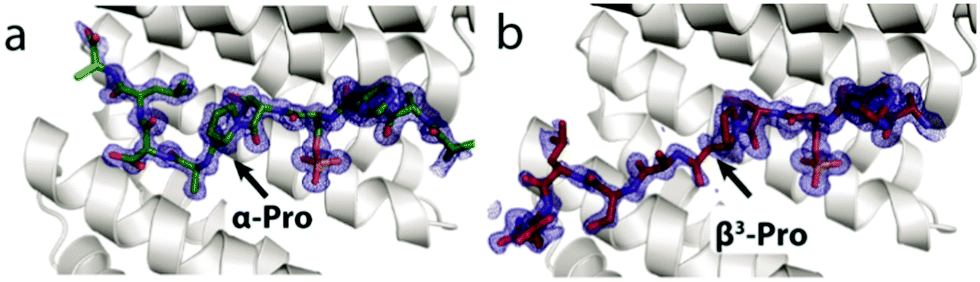 | ||
| Fig. 3 Crystal structures of YAP1.1 (a, green sticks, PDB 3MHR), βYAP2 (b, red sticks, PDB 6G6X). The secondary structure for the 14-3-3σΔC protein (grey cartoon) differs very little between the three co-crystal structures. | ||
In the case of the disubstituted α/β-peptides; βYAP2.2 introduces more rotational freedom and is slightly less active in FP compared to than βYAP2. βYAP2 plots a “middle road” between the two β3-proline conformations observed in the crystal structures of βYAP2 and βYAP2.2. For βYAP3.2 the β3-alanine and serine residues N-terminal to the phosphoserine can be observed, thus being slightly more structured in this region than βYAP3, but with similar activities observed in FP.
On comparison of the FP data (14-3-3ζ) for YAP1.1 (IC50 = 3,7 μM), YAP2.2 (IC50 = 0,84 μM), βYAP2 (IC50 = 61 μM) and βYAP4 (IC50 = 35 μM), to separate backbone from chain-end effects, the single α → β3 mutation leads to a significant decrease (up to 73-fold) in potency whereas modifications at the C- and N-termini exert only a marginal effect on the phosphopeptide binding affinity. Although the Kd values of YAP1.1 and YAP2.2 are similar, the thermodynamic parameters associated with these two α-phosphopeptides are significantly different (Fig. S1, ESI†). Acetylation of the N-terminus and C-terminal amidation (YAP1.1 → YAP2.2) removes two charges from the peptide, resulting in an increase in the enthalpic contribution (ΔH becomes more negative) with a corresponding decrease in the entropic contribution (−TΔS). The α → β3 mutation in βYAP2 results in a similar binding enthalpy as YAP2.2 but a higher Kd value due to a more positive entropic component. This trend suggests more flexibility in the amide backbone due to more degrees of rotational freedom introduced by the β3 amino acid residue. The point mutation disrupts the natural binding mode in the wt sequence (e.g.YAP1.1), initially disruptive, but introduces backbone flexibility, which enables access to a similar extended high energy binding contact/conformation with the 14-3-3ζ protein, as can be seen in the X-ray crystal structure (Fig. 3). Increased amide flexibility means though more energy that needs to be invested to fix the peptide in the active conformation. Such α → β3 mutational effects in PPIs, are different from effects observed on protein folding.26 The α → β3 mutation in βYAP4 does not lead to a net change in entropy but does disrupt the native binding mode (i.e. compared to YAP2.2) and associated favourable interactions. This can be observed in the corresponding X-ray structure by a decrease in the available electron density in the C-terminus of the α/β-peptide.
The observation that mutating the proline residue in the +2 position relative to the phosphoserine has a large influence on the peptide binding conformation and thermodynamics is also in line with earlier work that shows the evolutionary importance of this residue.17 The proline residue provides nature with a structural motif that allows the peptide to exit the 14-3-3 binding groove without paying a large entropic penalty. By adding more flexibility at this position through α → β3 mutation, the conserved structural motif is distorted with major implications for binding mode and thermodynamics. Indeed, of the binding peptides, the peptide bearing only a proline mutation, βYAP2, shows the largest decrease in binding affinity and a large shift in binding mode, underlining the structural importance of this residue in 14-3-3 binding interactions.
In conclusion this work reports the first example of α/β-peptides bound to 14-3-3 protein, and their co-crystal structures and, to the best of our knowledge, the first study on α,β-phosphopeptide inhibitors of PPIs. A number of single α → β mutants – βYAP5, βYAP6, βYAP7 – were found to be roughly equipotent with the wild-type α-peptide epitope, YAP2.2. Furthermore, mutating the +2 proline residue – i.e.βYAP2 – has a large influence on thermodynamic and structural binding properties, in line with earlier results on conserved 14-3-3 binding epitopes. The crystal structure data verifies the findings of the binding (FP/ITC) data and correlates with the loss in binding affinity and allows to construct a global picture of the impact of these α → β modifications. The increased flexibility introduced into the peptide backbone by the α → β mutations tended to decrease peptide binding due to changes in both the entropy and enthalpy of binding, which in some cases translated into well-defined alternative binding modes. These data provide yet further structural proof of the consequences of α → β modifications for the binding of α/β-peptide to their protein target, and their correlation to the biochemical and biophysical binding data.27–30 Such insights should help in the design of 14-3-3 PPI modulators and could be used to design peptides having multiple modifications to tune pharmacokinetic properties.
The authors would like to thank Pim de Vink for providing the FITC-ERα peptide for the FP studies. Funding was provided by the Netherlands Organization for Scientific Research (NWO) through Gravity program 024.001.035 and ECHO grant 717.014.001.
Conflicts of interest
There are no conflicts to declare.Notes and references
- Y. Aghazadeh and V. Papadopoulos, Drug Discovery Today, 2016, 21, 278–287 CrossRef CAS PubMed.
- A. Kaplan, C. Ottmann and A. E. Fournier, Pharmacol. Res., 2017, 125, 114–121 CrossRef CAS PubMed.
- K. L. Pennington, T. Y. Chan, M. P. Torres and J. L. Andersen, Oncogene, 2018, 37, 5587–5604 CrossRef CAS PubMed.
- L.-G. Milroy, L. Brunsveld and C. Ottmann, ACS Chem. Biol., 2013, 8, 27–35 CrossRef CAS PubMed.
- H. Fu, R. R. Subramanian and S. C. Masters, Annu. Rev. Pharmacol. Toxicol., 2000, 40, 617–647 CrossRef CAS.
- M. B. Yaffe and S. J. Smerdon, Annu. Rev. Biophys. Biomol. Struct., 2004, 33, 225–244 CrossRef CAS.
- C. Ottmann, S. Marco, N. Jaspert, C. Marcon, N. Schauer, M. Weyand, C. Vandermeeren, G. Duby, M. Boutry, A. Wittinghofer, J.-L. Rigaud and C. Oecking, Mol. Cell, 2007, 25, 427–440 CrossRef CAS.
- Y. Joo, B. Schumacher, I. Landrieu, M. Bartel, C. Smet-Nocca, A. Jang, H. S. Choi, N. L. Jeon, K.-A. Chang, H.-S. Kim, C. Ottmann and Y.-H. Suh, FASEB J., 2015, 29, 4133–4144 CrossRef CAS PubMed.
- I. J. De Vries-vanLeeuwen, D. da Costa Pereira, K. D. Flach, S. R. Piersma, C. Haase, D. Bier, Z. Yalcin, R. Michalides, K. A. Feenstra, C. R. Jiménez, T. F. A. de Greef, L. Brunsveld, C. Ottmann, W. Zwart and A. H. de Boer, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 8894–8899 CrossRef CAS PubMed.
- L.-G. Milroy, M. Bartel, M. A. Henen, S. Leysen, J. M. C. Adriaans, L. Brunsveld, I. Landrieu and C. Ottmann, Angew. Chem., Int. Ed., 2015, 54, 15720–15724 CrossRef CAS PubMed.
- L. M. Stevers, C. V. Lam, S. F. R. Leysen, F. A. Meijer, D. S. van Scheppingen, R. M. J. M. de Vries, G. W. Carlile, L. G. Milroy, D. Y. Thomas, L. Brunsveld and C. Ottmann, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, E1152–1161 CrossRef CAS PubMed.
- M. Würtele, C. Jelich-Ottmann, A. Wittinghofer and C. Oecking, EMBO J., 2003, 22, 987–994 CrossRef PubMed.
- M. Pelay-Gimeno, A. Glas, O. Koch and T. N. Grossmann, Angew. Chem., Int. Ed., 2015, 54, 8896–8927 CrossRef CAS PubMed.
- P. M. Cromm, K. Wallraven, A. Glas, D. Bier, A. Fürstner, C. Ottmann and T. N. Grossmann, ChemBioChem, 2016, 17, 1915–1919 CrossRef CAS PubMed.
- P. Thiel, L. Röglin, N. Meissner, S. Hennig, O. Kohlbacher and C. Ottmann, Chem. Commun., 2013, 49, 8468–8470 RSC.
- R. Rose, S. Erdmann, S. Bovens, A. Wolf, M. Rose, S. Hennig, H. Waldmann and C. Ottmann, Angew. Chem., Int. Ed., 2010, 49, 4129–4132 CrossRef CAS PubMed.
- M. B. Yaffe, K. Rittinger, S. Volinia, P. R. Caron, A. Aitken, H. Leffers, S. J. Gamblin, S. J. Smerdon and L. C. Cantley, Cell, 1997, 91, 961–971 CrossRef CAS.
- K. J. Peterson-Kaufman, H. S. Haase, M. D. Boersma, E. F. Lee, W. D. Fairlie and S. H. Gellman, ACS Chem. Biol., 2015, 10, 1667–1675 CrossRef CAS PubMed.
- I. Avan, C. D. Hall and A. R. Katritzky, Chem. Soc. Rev., 2014, 43, 3575–3594 RSC.
- W. S. Horne, M. D. Boersma, M. A. Windsor and S. H. Gellman, Angew. Chem., Int. Ed., 2008, 47, 2853–2856 CrossRef CAS.
- R. W. Cheloha, J. A. Sullivan, T. Wang, J. M. Sand, J. Sidney, A. Sette, M. E. Cook, M. Suresh and S. H. Gellman, ACS Chem. Biol., 2015, 10, 844–854 CrossRef CAS PubMed.
- J. W. Checco and S. H. Gellman, Curr. Opin. Struct. Biol., 2016, 39, 96–105 CrossRef CAS PubMed.
- R. W. Cheloha, A. W. Woodham, D. Bousbaine, T. Wang, S. Liu, J. Sidney, A. Sette, S. H. Gellman and H. L. Ploegh, J. Immunol., 2019, 203, 1619–1628 CrossRef CAS PubMed.
- B. Schumacher, M. Skwarczynska, R. Rose and C. Ottmann, Acta Crystallogr., Sect. F: Struct. Biol. Cryst. Commun., 2010, 66, 978–984 CrossRef CAS PubMed.
- T. Moroishi, C. G. Hansen and K.-L. Guan, Nat. Rev. Cancer, 2015, 15, 73–79 CrossRef CAS PubMed.
- Z. E. Reinert and W. S. Horne, Chem. Sci., 2014, 5, 3325–3330 RSC.
- E. F. Lee, B. J. Smith, W. S. Horne, K. N. Mayer, M. Evangelista, P. M. Colman, S. H. Gellman and W. D. Fairlie, ChemBioChem, 2011, 12, 2025–2032 CrossRef CAS PubMed.
- L. M. Johnson, W. S. Horne and S. H. Gellman, J. Am. Chem. Soc., 2011, 133, 10038–10041 CrossRef CAS.
- B. J. Smith, E. F. Lee, J. W. Checco, M. Evangelista, S. H. Gellman and W. D. Fairlie, ChemBioChem, 2013, 14, 1564–1572 CrossRef CAS PubMed.
- K. J. Peterson-Kaufman, H. S. Haase, M. D. Boersma, E. F. Lee, W. D. Fairlie and S. H. Gellman, ACS Chem. Biol., 2015, 10, 1667–1675 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9cc07982c |
| This journal is © The Royal Society of Chemistry 2019 |