
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Reprogramming biological peptides to combat infectious diseases
Marcelo
Der Torossian Torres
 and
Cesar
de la Fuente-Nunez
*
and
Cesar
de la Fuente-Nunez
*
Machine Biology Group, Departments of Psychiatry and Microbiology, Institute for Biomedical Informatics, Institute for Translational Medicine and Therapeutics, Perelman School of Medicine, and Department of Bioengineering, University of Pennsylvania, Philadelphia, Pennsylvania, USA. E-mail: cfuente@pennmedicine.upenn.edu
First published on 25th November 2019
Abstract
With the rapid spread of resistance among parasites and bacterial pathogens, antibiotic-resistant infections have drawn much attention worldwide. Consequently, there is an urgent need to develop new strategies to treat neglected diseases and drug-resistant infections. Here, we outline several new strategies that have been developed to counter pathogenic microorganisms by designing and constructing antimicrobial peptides (AMPs). In addition to traditional discovery and design mechanisms guided by chemical biology, synthetic biology and computationally-based approaches offer useful tools for the discovery and generation of bioactive peptides. We believe that the convergence of such fields, coupled with systematic experimentation in animal models, will help translate biological peptides into the clinic. The future of anti-infective therapeutics is headed towards specifically designed molecules whose form is driven by computer-based frameworks. These molecules are selective, stable, and active at therapeutic doses.
 Marcelo Der Torossian Torres | Marcelo Der Torossian Torres received his M.S. (2013) and PhD (2018) degrees in Science and Technology/Chemistry from Universidade Federal do ABC (Brazil) under the supervision of Prof. Vani Oliveira. In 2019, he joined the Machine Biology Group at the University of Pennsylvania as a post-doctoral researcher working with Prof. Cesar de la Fuente-Nunez. His research interests include the multifunctionality of bioactive peptides, the rational design of antimicrobial peptides by physicochemical-, structural- and dynamical-guided approaches, besides mechanisms of action and biophysics studies of biomolecules. |
 Cesar de la Fuente-Nunez | Cesar de la Fuente-Nunez is a Presidential Assistant Professor at the University of Pennsylvania, where he is leading the Machine Biology Group. Prof. de la Fuente is pioneering the computerization of biological systems, seeking to expand nature's repertoire to build novel synthetic molecular tools and devise therapies that nature has not previously discovered. The Machine Biology Group aims to develop computer-made tools and medicines that will replenish our current antibiotic arsenal. Prof. de la Fuente was named a Boston Latino 30 Under 30 (2016), a Top 10 MIT Technology Review Innovator Under 35 (Spain) (2016), a Wunderkind by STAT News (2018), a Top 10 Under 40 by GEN (2019) and is a recipient of the Society of Hispanic Professional Engineers Young Investigator Award (2019) and the Langer Prize (2019). He has also been recognized in 2019 by MIT Technology Review as one of the world's top innovators for “digitizing evolution to make better antibiotics”. His scientific discoveries have yielded over 70 peer-reviewed publications and multiple patents. |
Introduction
The rise of multi-resistant microorganisms has created a need for new strategies compared to conventional antibiotics.1 One alternative is antimicrobial peptides (AMPs), the most well studied class of bioactive peptides. These versatile molecules act through multiple mechanisms against pathogenic microorganisms and can have synergistic effects when combined with other families of antibiotics.2Peptides that have antimicrobial,3–5 antibiofilm,6,7 and immunomodulatory activities8,9 may be part of host-defense systems10 or designed in silico.11,12 These peptides exhibit common features such as small lengths,13 net positive charges,3–5 amphipathic structures,13 and broad-spectrum biological activities.7 However, recently there have been reported mechanisms of action revealing their specific extra- and intracellular targets.14
There are countless families and various structural compositions of AMPs, so standardized studies may not uniformly be the most appropriate way to improve the design and predict the activity of this class of molecules. Several attempts to increase the accuracy of AMP design for optimal activity and selectivity towards microorganisms have been reported recently.15,16 Moreover, AMPs, serving as novel antibiotics, act through diverse mechanisms,17 making it difficult for microorganisms to acquire resistance.18,19
Among the prospective antibiotic candidates, the ones aimed at precise treatment or prevention stand out as promising alternatives that could help overcome the challenge imposed by multidrug-resistant microorganisms.1 Nucleic acid-based systems, such as the clustered regularly interspaced short palindromic repeats-associated systems (CRISPR-Cas), have been recently used to generate sequence-specific antimicrobials, in addition to their applications for precise RNA-guided genome editing, epigenetic modification, and gene regulation in numerous eukaryotic and prokaryotic organisms.20 Another example of precise treatment is the use of peptide nucleic acids, i.e., synthetic polymers composed of N-(2-aminoethyl)-glycine units and purine or pyrimidine bases linked by peptide bonds. Peptide nucleic acids act by inhibiting the translation of target genes.21
Despite the efforts and advances made in the peptide field over the last decades, the characteristics of AMPs that directly affect their activity are still not completely understood. Peptide researchers usually categorize the main physicochemical and structural features of these complex molecules by correlating them with biological activities.22,23
The activity determinants that might be extracted experimentally are less complex yet directly related to the most fundamental physicochemical properties of the peptides. Hydrophobicity-related properties, net charge, and helicity are among the most often used features for the design of AMPs. Mutagenesis of known molecules can reveal how changes in these properties affect activity. Progress in bioinformatics and computational biology has led to the development of more complex and descriptive features related not only to physicochemical properties but also to structural features at a microscopic level.22,23
This Feature Article reports recent efforts to design and engineer peptides as novel anti-infective agents. We envision that the applications of AMPs go beyond the generation of antibiotics. Understanding their most basic features from first principles, at a level that allows us to predict their function, might be the key for all protein/peptide involved processes, precipitating their application in materials science and medical devices and leading to ground-truth understanding of fundamental physicochemical properties, such as protein folding, self-assembly, and the effects of diverse organic materials.
Discovery of biologically active peptides in nature
Before the introduction of computational biology techniques, there were fundamentally two ways to obtain biologically active peptides: isolating natural peptides from living beings or through template-based design of new peptides (Fig. 1). Isolation is typically followed by purification, characterization, and screening for the biological purposes desired. The design of template-based peptides is based on exhaustive structural analyses of previously described bioactive peptides that are subsequently synthesized and screened for their biological activity; the aim is to obtain “hits” that can be explored as new templates for another round of template-based design.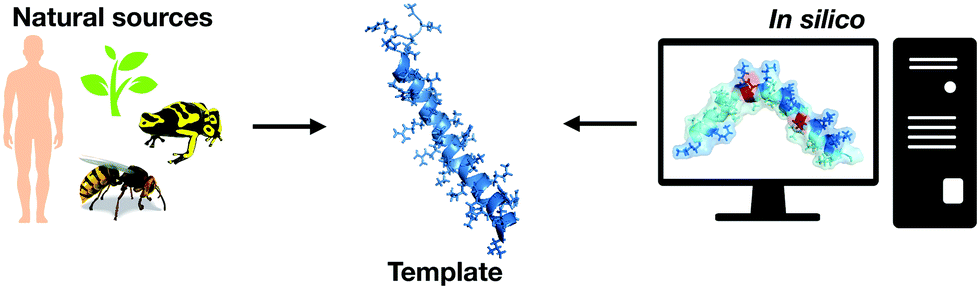 | ||
| Fig. 1 Antimicrobial peptides might be obtained from diverse natural sources, such as insects, arachnids, mammalians, plants, and microorganisms. In silico generation of peptides is also an important source of prospective synthetic antimicrobial compounds. | ||
However, the isolation of natural molecules and the template-based design of new peptides are costly and time-consuming approaches with a low success rate. Computational biology tools, along with high throughput screening methods, have brought biophysics, physical-chemistry, and chemical biology derived complexity to the design of bioactive peptides, generating an extraordinary growth in data relative to that previously available from sequences deposited in databases. This vast increase in data subsequently led to the need for refined ways to transform it into useful information for the design and optimization of active peptides. Data mining this information manually is virtually impossible, since the sequence space of peptides is extremely large, for instance, a 15 residues peptide would have a total sequence space of 2015 (3.2768 × 1019 sequences) considering only the natural amino acids. Thus, computational tools dedicated to refining data analyses have been extensively adopted for the purpose of drug design.12
Identification of cryptic peptides from natural templates
Among the most promising computational biology tools for the discovery of functional peptides, pattern recognition algorithms (PRAs) stand out because of their accuracy and usefulness. PRAs enable comparative modeling, that is, the identification of encrypted templates from the proteolytic processing of large precursors that do not necessarily present biological activity (Fig. 2).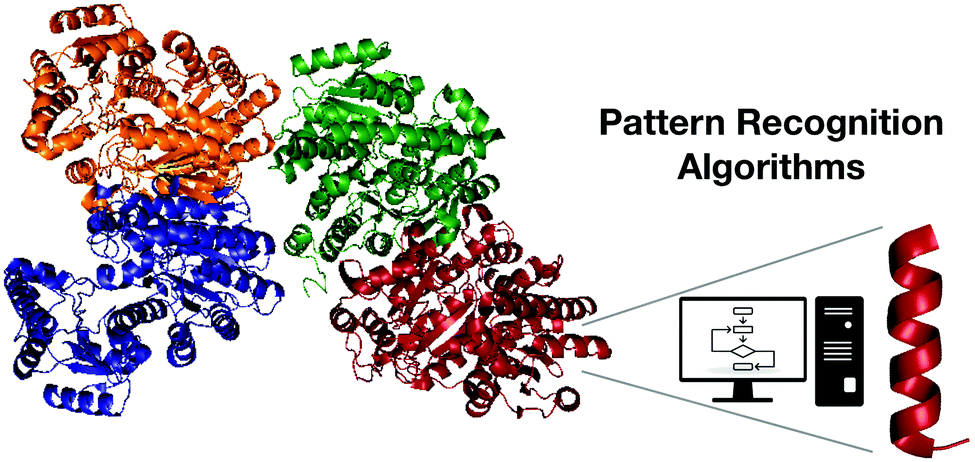 | ||
| Fig. 2 Pattern recognition algorithms as a useful tool for discovering encrypted active fragments from larger peptides or proteins. | ||
Typically, the algorithms account for well-established chemical, physical, or biological parameters that are known to influence biological activity. For instance, Pane et al.24 reported the linear correlation of antimicrobial activity to the distribution of net charge, hydrophobicity, and the length of the sequences, considering the relative contribution of each parameter defined by the algorithm in a strain-specific manner. The fitness function allowed the identification of AMPs through a computational-experimental framework with which the scoring functions of the activation peptide of pepsin A, the main human stomach protease, and its N- and C-terminal halves were also reported as AMPs.25 The three peptides from pepsinogen A3 isoform, (P)PAP-A3 (PIMYKVPLIRKKSLRRTLSERGLLKDFLKKHNLNPARKYFPQWAPTL), (P)IMY25 (PIMYKVPLIRKKSLRRTLSERGLLKD) and FLK22 (FLKKHNLNPARKYFPQWAPTL), were prepared in a recombinant form using a fusion carrier specifically developed to express toxic peptides in Escherichia coli.26 Recombinant pepsinogen A3-derived peptides presented wide-spectrum antimicrobial activities, with MIC values in the range 1.56–50 μmol L−1, and no toxicity toward human cells, and they exhibited anti-infective activity in vivo. Moreover, the activation peptide was bactericidal at pH 3.5, which is relevant to foodborne pathogens. The authors show that this new class of previously unexplored AMPs contributes to microbial surveillance within the human stomach.
Bioprospection of anti-infective peptides
Several approaches for the prediction of active peptides from natural templates have been reported over the last decades.27 Bioinformatics tools have contributed substantially to unravelling the role of descriptors in structure–activity studies and natural motifs leading to important biological functions28 (Fig. 3). The understanding of the role of descriptors enables not only the rational design of peptides but the prediction of their biological functions.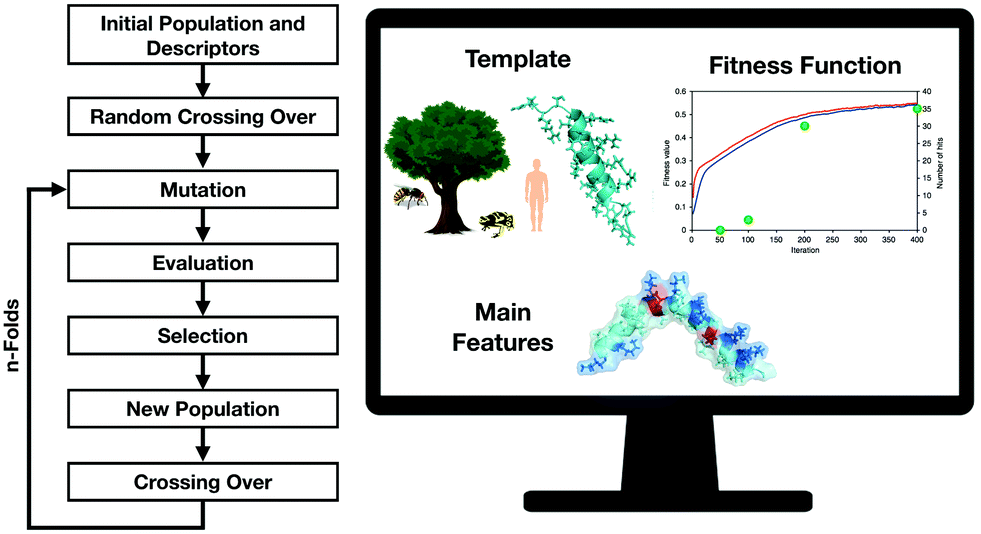 | ||
| Fig. 3 Genetic algorithms are versatile tools for the design of new-to-nature peptides based on the analysis of molecular descriptors variations after the proposed mutations followed by selection and generation of the new population. Adapted from Porto et al.29 | ||
The use of computer-aided design of synthetic derivatives from natural templates represents a novel strategy for the generation of active peptides.30–32 For example, the guava fruit-derived peptide Pg-AMP1(RESPSSRMECYEQAERYGYGGYGGGRYGGGYGSGRGQPVGQGVERSHDDNRNQPR) has been used as a template to generate the guavanin synthetic peptides by means of a genetic algorithm (GA) that introduces specific modifications. The methodology included a descriptive equation as a fitness function that drives the algorithm, and the interruption of the algorithm before reaching a diversity plateau, thus allowing exploration of another parcel of peptide combinatorial sequence space containing completely different peptide sequences. The approach was based on the most well-known physicochemical and biophysical properties of AMPs such as hydrophobicity, net charge, and polar/nonpolar ratio. Among the new computer-generated sequences, perhaps the most interesting is the synthetic peptide guavanin 2 (RQYMRQIEQALRYGYRISRR), which is bactericidal at low micromolar concentrations. The mechanism of action of guavanin 2 was revealed to involve disruption and hyperpolarization of membranes in E. coli cells.29
The greatest advantage of using GAs is the flexibility of exploring any property of the molecules. Free energy has been studied for several smaller organic compounds;33 however, very few thorough energetic studies have been done for peptides and proteins, as the complexity of those systems is computationally costly and time-consuming. Supady et al. reported the identification of low-energy conformers employing a GA that searched segments of the conformation space for molecules with lower energetic profiles. The authors aimed to predict all conformers within an energy window above the global minimum instead of finding the global minimum energy. They used well-established first principles of molecular structure for drug design to evaluate a data set extracted from a database consisting of amino acid dipeptide conformers and compared the performance of a systematic search with that of a random conformer generator.33
The massive data collections generated by high-throughput screens require effective collecting, interpreting and integrating techniques.34 Machine learning (ML) is one of the most active areas of research in computer science, with broad applications in the fields of biological engineering and synthetic biology (Fig. 4) that allow the interpretation and integration of high-throughtput data.35 Among the numerous applications of this methodology, the efficient optimization of antimicrobial compounds is of particular interest for combating the growth of antimicrobial resistance. Yoshida et al. presented a proof-of-concept methodology for efficiently optimizing the efficacy of AMPs.36 The authors combined a GA, ML, and in vitro evaluation to create optimal peptide candidates against E. coli. Forty-four lead compounds were identified, all of them derived from a small cationic natural template. The hits obtained were up to 160-fold more active than the wild-type peptide after three cycles of predictions. This technique also enabled the authors to design peptides with different structural tendencies, ranging from unstructured to well-defined helical molecules, which were more active than the wild-type. Results such as those obtained by Supady et al. exemplify how ML can provide tools and accelerate the discovery of AMPs with promising antimicrobial activities, allowing the exploration of structural patterns and indicating how particular descriptors influence biological activity.
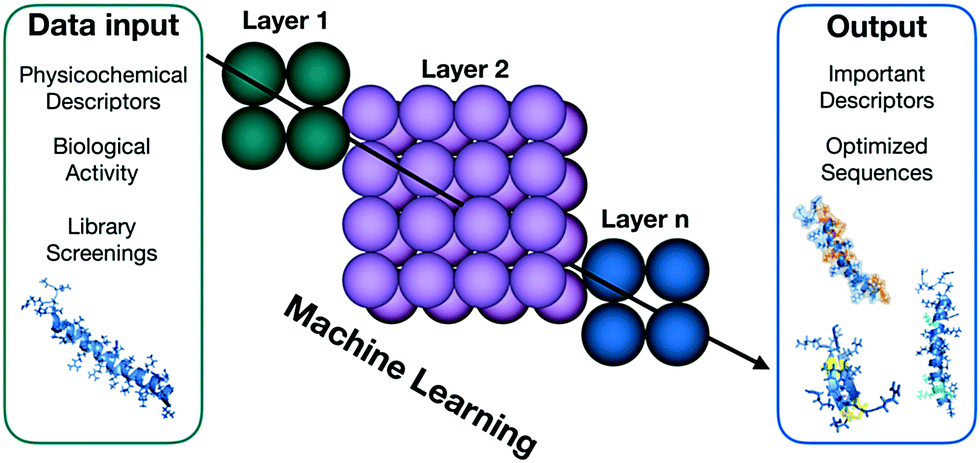 | ||
| Fig. 4 Machine learning uses statistical methods to enable computer systems to learn and progressively improve their performance as far as generating bioactive compounds from physicochemical, structural and biological activity data. | ||
Mechanisms of action also fall within the compass of machine learning-based design models. Lee et al. developed a support vector machine-based classifier to investigate α-helical AMPs with activity in bacterial membranes.37 The model related α-helicity, determined by X-ray scattering, to in vitro antimicrobial activity. The authors were able to associate negative Gaussian membrane curvature as an indirect measure of antimicrobial activity.
Maybe the most promising of the artificial intelligence design techniques is the hierarchically embedded neural networks, also known as deep learning (DL).38 The main advantage of DL over other approaches, such as GA and ML, is the direct prediction of peptides antimicrobial activity intead of relying on the calculation of intrinsic or other complex structural and physicochemical properties of these molecules. However, this advantage might also be an disadvantage, since the access to reliable and comparable biological data in large amounts is difficult because we currently lack standard procedures for purifying and testing peptides. Muller et al.39 proposed using long short-term memory recurrent neural networks for designing combinatorial de novo peptides. The model learned from patterns present in α-helical AMP sequences from databases and was able to generate new peptides from the learned context. The authors reported that most of the AMPs generated (82%) were indeed active.
Design approaches to improve on biology's templates: the advent of synthetic and computer-made peptides
The rise of multi-drug resistant microorganisms has led to a post-antibiotic era: conventional antibiotics have lost their effectiveness and infections caused by antibiotic-resistant organisms lead to serious health problems and can be lethal. As a result, it is essential that new alternatives with potential to fight these resistant infectious agents are developed. AMPs are promising alternatives to classical antibiotics, as they present diverse mechanisms of action;16,28 sometimes, AMPs are able to slow down the evolution of resistance.2Despite many efforts, structure–activity relationship (SAR) studies are not conclusive regarding the impact of physicochemical properties and structure on biological activities, mostly because of the lack of standard procedures for accurately determining descriptor values and experimental protocols.
Currently, SAR studies, by indicating ways to systematically modify the peptides, can be used to determine how changes in composition and structure affect biological activities. The overall aim comprises maximizing antimicrobial activity and resistance to proteolytic degradation, while minimizing toxicity towards the host. There are several ways to classify the most commonly used design techniques. The most well-known methodologies that have been used to design new AMPs and guide SAR studies are site-directed mutagenesis, computational design approaches, synthetic libraries, template-assisted methodologies, and mechanism-based strategies (Fig. 5).
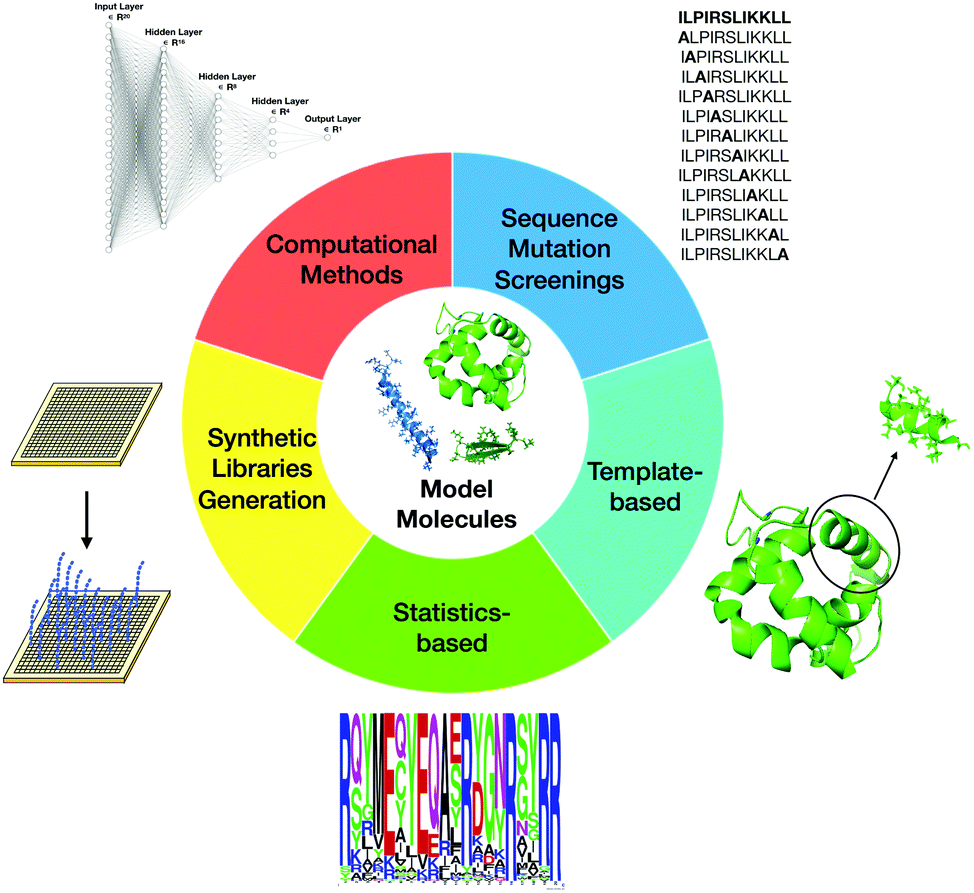 | ||
| Fig. 5 Main design strategies used for repurposing toxic peptides into potent antimicrobial agents. | ||
Mutagenesis
There are several effective ways to design and evaluate a family of peptides by means of substituting amino acid residues in the original sequence by residues with different properties.28Physicochemical-guided peptide design is the most effective approach for also considering the effects of specific changes made to the structure of AMPs (Fig. 6). For example, polybia-CP (ILGTILGLLKSL-NH2), a toxic wasp venom peptide, was redesigned to be a non-toxic antimicrobial agent with anti-infective activity in vivo through the systematic evaluation of each residue of the original sequence. First, an Ala-scan screening was performed to check the role of each residue in structure and biological activities. The information obtained allowed the design of a second generation of polybia-CP derivatives with substitutions that directly influenced the antimicrobial and cytotoxic activities while the helical structure of the peptides was favored. The strategy supported helicity as the most important structural feature of the antimicrobial activity of these small cationic amphipathic peptides, while hydrophobicity-related properties were directly responsible for the toxic activity (Fig. 6).40
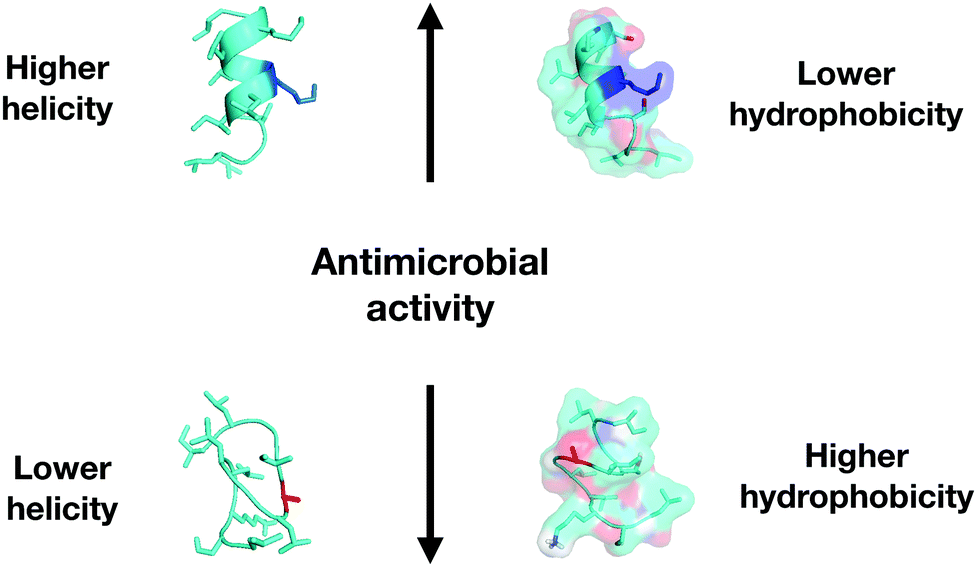 | ||
| Fig. 6 Correlation of physicochemical descriptors and antimicrobial activity for polybia-CP and its enhanced derivatives. | ||
Using a similar approach, we obtained different results for decoralin, a small cationic amphipathic peptide derived from wasp venom. Decoralin (SLLSLIRKLIT-NH2) Ala-scan screening revealed the importance of a Lys residue for the biological activities of the peptide, and changes on the hydrophobic face of the amphipathic structure led to higher helical tendency and increased resistance to degradation by proteases. The helicity of the decoralin family did not correlate to activity, although the balance between the increased hydrophobicity and the insertion of positively charged residues on the hydrophilic face and on the interface of the helical structure generated highly active peptides.41,42 The decoralin derivatives also inhibited the growth of MCF-7 human breast cancer cells43 and Plasmodium sporozoites.44
High-throughput peptide synthesis, by enabling the correlation of the most important biological descriptors with the biological activities of the peptides,45 is another powerful tool for mutagenesis studies.45,46 The systematic substitution within a native sequence of all possible options for each amino acid generates a synthetic library and a large amount of data that can guide the design of peptides with enhanced activity. It is not possible to evaluate the entire sequence space of the peptides; however, representative canonical residues of each kind of amino acid (basic, acidic, aliphatic, hydrophobic, and pseudo-amino acid) are chosen first to explore accentuated differences in biological and physicochemical descriptors45 (Fig. 7).
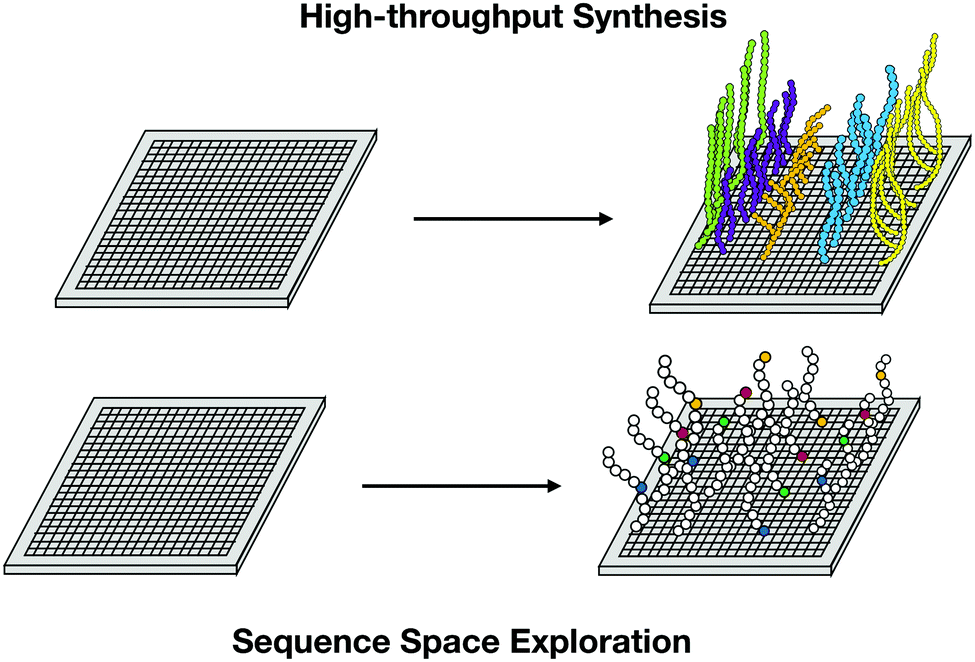 | ||
| Fig. 7 High-throughput synthesis allows massive data collection and exploration of peptides sequence space for the analysis of relevant descriptors contributing to biological function. | ||
The use of D-enantiomeric amino acids for mutagenesis studies is a well-known approach for preventing peptide degradation in the presence of proteolytic enzymes.44D-Enantiomeric amino acids have greater bioavailability than L-enantiomers, and their use makes it possible to avoid abrupt changes in structural and physicochemical properties, which may have unforeseen functional effects. Thus, peptides consisting of D-amino acids are powerful tools for mutagenesis studies. For instance, we proposed the design of a series of peptides including L-, D-, and retro-inverso peptides. In this series, the D-peptides presented the most promising low cytotoxicity against mammalian cells, which is an expected effect of the insertion of D-amino acids on lytic peptides sequences,47 and inhibitory activity at low concentrations (10 μg mL−1) against the formation of biofilms by multidrug-resistant P. aeruginosa strains.48,49 Biofilms are aggregates of microorganisms in which cells are embedded in a self-produced matrix of extracellular polymeric substances that are adhere to each other and to a surface50 (Fig. 8A).
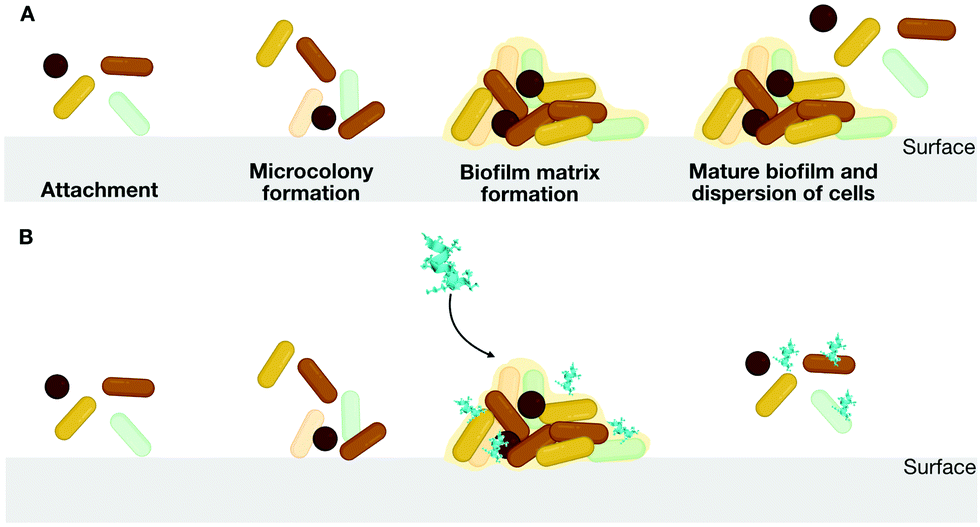 | ||
| Fig. 8 (A) Biofilm formation from the attachment of cells to the biofilm surface to colonization and maturation of the colonies. The process is interrupted by (B) the addition of antibiofilm peptides. | ||
Di Grazia et al.51 showed the effect of rational substitutions of L-amino acids by D-amino acids in a very comprehensive study about esculetin-1a(1–12) (GIFSKLAGKKIKNLLISGLKG-NH2). The authors showed that compared to the wild-type, the D-peptide derivative was significantly less toxic towards mammalian cells, more effective against P. aeruginosa biofilms, more stable in serum, and had increased ability to promote migration of lung epithelial cells.
Mutagenesis studies must always contain molecular descriptors, which are physicochemical or structural-related parameters that describe which peptide sequences correspond to particular biological functions. These descriptors can have multiple levels of complexity, making it difficult to attribute to a specific descriptor its actual effect, or weight, on the biological functions shown by the peptides. There are several characteristics of the sequences that are responsible for their descriptor values such as molecular weight, structure, volume, number of rotatable bounds, atom types, electronegativities, polarizabilities, topological charge, among several others. The majority of these descriptors are non-empirical and generated through knowledge-based, graph-theoretical, molecular mechanical or quantum-mechanical tools. The descriptors are classified according to scalar physicochemical or structural features; bi-dimensional and three-dimensional molecular descriptors are classified according to their amino acid sequences.32
A solid theory that can model and describe all sorts of structural and physicochemical properties of AMPs would rely on the standardization of experimental protocols, which is so far lacking. The absence of standardization is, in fact, the major obstacle to applying the results described in mutagenesis studies to clinical studies. It is difficult to discern a rational design set of rules among experimental procedures that are different from other studies in the literature. Advances in computational technologies applied to the design of peptides are starting to provide a basis for more clear explanations and very detailed design processes that might be used to design similar classes or families of bioactive peptides.12,26,29,31,32 However, simpler approaches, such as homology by alignment, structure–function design, and high-throughput screens, can also provide standardized datasets that are easy to compare. Mardirossian et al. described Tur1A (RRIRFRPPYLPRPGRRPRFPPPFPIPRIPRIP), a proline-rich AMP from bottleneck dolphins that is internalized by bacterial cells and acts, in E. coli, by targeting the bacterial ribosome with low cytotoxicity toward mammalian cells. This peptide was identified after comparison with orthologous mammalian proline-rich host-defense peptides that present the same mechanism of action.52 Another example is structure–function-guided design, which involves systematic substitutions made to the sequence of a particular template molecule. For the wasp venom AMP polybia-CP, we first generated analogs with increased activity by fine-tuning modifications in specific residues of the helical structure that would confer higher helical tendency and then made small modifications to decrease the affinity of the peptides to mammalian cells, generating selective AMPs with low cytotoxicity and high antimicrobial potency.40
In silico combinatorial exploration: towards the design of artificial, computer-made therapeutic peptides
Several active peptides have been generated by combinatorial design based on activity descriptors that do not account for structural comparison with existing molecules but, rather, are based on known physicochemical properties of the amino acid residues present in the sequences of the designed peptides. Essentially, the de novo-designed AMPs are restricted to pre-established motifs that consider amphipathic balances between polar and hydrophobic residues and sequence length explorations.53 Optimization of de novo-generated AMPs via the use of GAs has yielded potent antimicrobial agents.54,55 However, some of the resulting peptides might also be toxic because some of the properties neglected for their design, such as hydrophobic-related features, lead to high affinity towards eukaryotic membranes.56 Design strategies are now taking into consideration more complex descriptors, and the AMPs generated de novo are more selective for prokaryotic cells,39,57,58 not only for antimicrobial purposes but for other biochemical processes, such as peptide substrates for enzymes59 that affect essential biochemical pathways of prokaryotic microorganisms. The in silico exploration have allowed to expand the usefulness of peptides for other technological uses.Antibiofilm peptides
The formation of pathogenic bacterial biofilms on medical devices and damaged body tissues is a challenging obstacle to the effective treatment of bacterial infections in clinical settings.60 The steady increase in the number of drug-resistant microorganisms forming biofilms during treatment or post-surgery highlights the need to effectively combat biofilms (Fig. 8B). We have reported several classes of AMPs as versatile antibiofilm agents.48,61–71LL-37 (LLGDFFRKSKEKIGKEFKRIVQRIKDFLRNLVPRTES) and its fragment sequence 1037 (RFRIRVRV-NH2) were two of the first AMPs described as antibiofilm peptides against P. aeruginosa laboratory strains under dynamic continuous flow conditions72 and also against pathogens isolated from cystic fibrosis patients.61 Later, the bactenecin-derived AMP 1018 (RLIVAVRIWRR-NH2) and additional analogs were also described as antibiofilm peptides with higher activity against biofilms than planktonic bacteria, and more active than other active molecules, such as LL-37 and 1037, under the same conditions. These results revealed potentially different mechanisms underlying peptide-mediated targeting of planktonic versus biofilm cells. The hydrophobicity-related features of this family of peptides played an important role in their antibiofilm activity. Peptide 1018 and its analogs with conserved or increased hydrophobicity were able to eradicate 99% of the biofilm and even prevent biofilm formation61 and did not increase their toxicity.
Another peptide described as antibiofilm agent6 include DJK-5 (vqwrairvrvir-NH2), a D-enantiomeric synthetic analog of a previously described antibiofilm peptide (1018). DJK-5 is still among one of the most active molecules against bacterial biofilms and effectively targeted several species, such as P. aeruginosa, E. coli, Acinetobacter baumannii, Klebsiella. pneumoniae, and Salmonella enterica. DJK-5 is able to eradicate and prevent biofilm formation at 0.5–8 μg mL−1, a concentration range lower than that at which LL-37 is effective (∼50% inhibition at 16 μg mL−1).72 DJK-5, which is non-cytotoxic, also has the advantage of being resistant to proteolytic degradation and consequently, showing activity in animal models.
Random peptide mixtures73 or copolymers74,75 that mimic peptides have been described as a potential alternative to mutagenesis studies for generating antimicrobial and antibiofilm agents.76,77 For instance, Stern et al. used sequence random hydrophobic-cationic peptides formed by L-phenylalanine and L-/D-lysine mixtures against methicillin-resistant Staphylococcus aureus biofilms. The homochiral and heterochiral 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio mixtures presented MICs of 6 μg mL−1, acting through penetrating the bacterial cells, low hemolytic activity and were more effective at eradicating biofilms than the antibiotic daptomycin.
:1 ratio mixtures presented MICs of 6 μg mL−1, acting through penetrating the bacterial cells, low hemolytic activity and were more effective at eradicating biofilms than the antibiotic daptomycin.
Conjugating peptides with other molecules, such as lipids and antibiotics is also a promising strategy for increasing their antimicrobial and antibiofilm activities. For instance, Bionda et al.78 designed synthetic cyclic lipopeptide analogs of fusaricidin through a positional scanning combinatorial approach. Some of the analogs proposed by the authors were able to inhibit ESKAPE pathogens growth and formation of P. aeruginosa and S. aureus biofilms.
Immunomodulatory peptides
The role played by AMPs goes beyond their known activities against pathogenic bacteria. As naturally occurring AMPs evolved over millions of years, they became essential components of the innate immune system.79 Immunomodulatory AMPs act in innate immunity through complex immunomodulatory pathways, many of which are still unclear.80We recently described high-throughput screening methods for assessing the diverse immunomodulatory activities of peptides. These methods can be applied to design new synthetic immunomodulators. For instance, these methods generated innate defense regulator peptides (IDR) synthetic analogs that, besides presenting high activity against methicillin-resistant Staphylococcus aureus, also stimulate the production of monocyte chemoattractant protein (MCP-1) and suppress LPS-induced interleukin (IL)-1β production in human peripheral blood mononuclear cells.81 These peptides showed immunomodulatory activity profiles similar to that of the potent peptide 1018.82 Additionally, 1018 presented activity against P. aeruginosa and Burkholderia cenocepacia cystic fibrosis isolates.61,62
Another example of engineered synthetic peptides with varied immunomodulatory activities is clavanin-MO (FLPIIVFQFLGKIIHHVGNFVHGFSHVF-NH2), which modulates innate immunity by stimulating both leukocyte recruitment to the site of infection, and the production of immune mediators GM-CSF, IFN-γ and MCP-1. Clavanin-MO also suppresses the inflammatory response, preventing it from becoming excessive and potentially harmful, by increasing the synthesis of anti-inflammatory cytokines, such as IL-10, and repressing the levels of the pro-inflammatory cytokines IL-12 and TNF-α.83
Lipopeptides, such as amphomycins, polymyxins, teicoplanins, and bacitracins,84 are also well-known for their varied mechanisms of action, which includes intracellular targets85 and immunomodulatory activity.86 For example, daptomycin has immunomodulatory properties, resulting in the suppression of cytokine expression after host immune response stimulation by methicillin-resistant S. aureus. Although daptomycin is structurally related to amphomycins, which inhibit peptidoglycan biosynthesis, daptomycin was shown to be a membrane permeabilizing lipopeptide that is also capable of depolarizing the bacterial cell membrane.
Antimicrobial peptides as antibiotic potentiators
Exploiting the synergistic effects of antimicrobial agents that act through different mechanisms is being explored as a promising approach to the problem of antimicrobial resistance. Molecules such as AMPs, which sometimes present different mechanisms of action against certain microorganisms, are versatile tools to increase susceptibility and re-sensitize antibiotic-resistant microorganisms.For example, the antimicrobial and potent antibiofilm peptide 1018 has been reported to synergize with ceftazidime, ciprofloxacin, imipenem, and tobramycin, decreasing by 2- to 64-fold the concentration of antibiotic required to treat biofilms formed by P. aeruginosa, E. coli, A. baumannii, K. pneumoniae, S. enterica, and methicillin-resistant S. aureus,70 which represent most of the multidrug-resistant ESKAPE pathogens included in a list of multidrug-resistant bacteria created by the World Health Organization (Enterococcus faecium, S. aureus, K. pneumoniae, A. baumannii, P. aeruginosa, and Enterobacter spp.). Other examples of AMPs that synergize with antibiotics are the potent and stable synthetic peptides DJK-5 and DJK-6 (vqwrrirvwvir-NH2). Their activity against multidrug-resistant carbapenemase-producing K. pneumoniae isolates led to 16-fold lower concentrations of β-lactamases, such as meropenem, needed to combat 2-day-old biofilms.63
The role of AMPs as re-sensitizers of resistant bacteria has been systematically evaluated by Lazar et al. By screening the effect of 24 AMPs against 60 resistant E. coli strains, the authors found that the antibiotic-resistant bacteria presented a high frequency of collateral sensitivity to AMPs but very little cross-resistance. The authors also identified clinically relevant multidrug-resistance mutations that led to increased bacterial sensitivity to AMPs and described regulatory changes shaping the lipopolysaccharide composition of the bacterial outer membrane that were directly related to the collateral sensitivity in these multidrug-resistant microorganisms.2 Therefore, AMP-antibiotic combinations may be able to enhance antibiotic activities against multidrug-resistant bacteria, and most likely those combinations will slow down the de novo evolution of resistance.
In vivo testing of synthetic bioactive peptides
Only a small number of biologically active peptides is currently being used in the clinic, such as such as polymyxin B, gramicidin S, nisin, caspofungin, and brilacidin, and in clinical trials.87,88 To reach this stage, the AMPs have to show potential for overcoming all existing limitations and bioavailability issues and show efficacy in a range of conditions using standardize experiments. As examples of peptides in advanced phases (phase III) of clinical trials we highlight D2A21, a synthetic peptide for the treatment of infected burns and wounds that operates by interacting directly with the membrane of the pathogens.89 Additionally, SGX942 is small synthetic cationic AMP, for the treatment of oral mucositis through multiple mechanisms of action, ranging from disrupting bacterial cell membrane to long-lasting immunomodulatory effects.90Peptide stability and bioavailability have limited their use, but the most important limitation is the lack of in vivo studies, as assessing their efficiency in vivo is essential. Animal models are the closest approximations to clinical conditions and the most reliable manner of assessing the efficacy of peptides and delivery methods. Rodents are the most frequently used class of animals for peptide studies in vivo, because they exhibit similarity to the relevant clinical conditions and because these animals are readily available, cheaper than other animals such as rabbits, pigs and non-human primates, and easy to handle. A considerable number of animal models aim to mimic conditions ranging from simulating biofilm and systemic bacterial infections to neurodegenerative or complex disorders caused by microorganisms. The reproducibility and translatability of in vitro bioactivity results into in vivo assays is still a great challenge because the in vitro conditions are not ideal and far from comparable to the ones encountered by the molecules in animal models. An alternative to this problem is simplifying the animal models to screen for candidates for subsequent, more complex tests. We have experienced diverse behavior of peptides when translating these active molecules from in vitro antimicrobial and antibiofilm assays to skin scarification mouse models. Usually, peptides present lower activity in vivo due to the lack of stability or the multitude of interactions that they undergo with the multitude of different classes of molecules in a living animal. To minimize these obstacles we have decided to test peptides in skin scarification murine models of abcess formation.25,29,40,91,92 The computationally generated peptide, EcDBS1R5 (PMKKLKLALRLAAKIAPVW), inspired by an E. coli AMP, was active at 8 μmol L−1 against P. aeruginosa in in vitro assays, while in a skin scarification mouse model infected by P. aeruginosa, the same peptide was active only at a concentration 8-fold higher (64 μmol L−1),92 even though the concentration was lower than its cytotoxic activity (>128 μmol L−1). Another example with the same translational behavior is PaDBS1R6 (PMARNKKLLKKLRLKIAFK), a synthetic peptide designed by the Joker algorithm. PaDBS1R6 is an AMP selective for Gram-negative bacteria that was not active against mammalian cells (>128 μmol L−1). It presented in vitro activity against P. aeruginosa at 8–16 μmol L−1, while its activity in animal models was shown at 64 μmol L−1.91 However, in some cases, we observed peptides that were as active in vivo as they were in vitro, such as polybia-CP and its analogs, which presented anti-P. aeruginosa activity at 4 μmol L−1.40 Additional technology development and modelling will be required to ensure predictive correlations between data acquired in vitro and that obtained in vivo.
Synthetic biology platforms for antimicrobial peptide production
Despite the significant progress in studies involving peptides and small proteins, the large-scale production of these molecules with high yields and purity for further commercial applications remains challenging.28Mostly, AMPs are extracted from natural sources or synthesized chemically. Isolation and purification of AMPs from natural sources are very useful tools for initial screenings; however, the overall process is extremely laborious and results in low yields. As an alternative, solid-phase peptide synthesis has been exhaustively used and optimized in the last decades and is now very effective for all kinds of peptides, including complex cyclic peptides and non-canonical amino acid-containing peptides. However, solid-phase peptide synthesis is still limited to peptides containing more than 35–40 residues.93 Coupling and deprotection steps are less effective in peptides this long,94 and there are side-reactions and cross-reactions that are difficult to avoid.94 Moreover, volumetric use of toxic reagents for amino acid coupling and/or activation reaction increases with the number of peptide chain residues, posing adverse environment impacts. An alternative is the SPOT synthesis of peptides, which enables rapid synthesis and screens using peptides at small scales. However, the yield and purity are still an issue.95
Synthetic biology approaches of recombining DNA offer a more sustainable, scalable, and cost-effective production of AMPs than the chemical-synthesis route because synthetic biology offers an unusually high degree of flexibility for genetically engineering microorganisms such as bacteria and yeasts.96 The heterologous expression of AMPs is generally performed by expressing a fusion protein to facilitate microbial production as well as simplified purification, decreasing toxicity to the producer cell and increasing resistance to enzymatic degradation. Usually, the carrier proteins are then cleaved and separated from AMPs.97 Deng et al. reviewed different types of cell factories used to produce AMPs, including E. coli and Bacillus subtilis and the yeast Pichia pastoris and Saccharomyces cerevisae.98
Based on the physicochemical and structural properties of AMPs, such as cationicity, amphipathicity, and hydrophobic-related features, AMPs are highly active against several host organisms. The selection of microorganisms and the rational design of plasmid libraries have led to the production of functional recombinant AMPs with improved expression yields. However, there are no general rules for selecting expression hosts, plasmid features, and fusion tags for producing a given fusion protein-AMP with guaranteed maximum productivity. Therefore, it is important that expression is conducted using well-known host microorganisms and plasmid libraries, and well-established approaches.99
As an alternative to the well-stablished E. coli and cell free extracts platforms for AMP production, we have engineered a versatile, on-demand yeast-based synthetic biology platform that allows AMP production in bioreactors.100,101 Using this technology, we were able to express the AMP apidaecin-1b (GNNRPVYIPQPRPPHPRL) at higher yields and lower cost than those obtained using E. coli-based platforms. The AMP produced had the same biological activities in vitro and in vivo when compared to both its chemically synthesized and purified version.102
The future of peptides as anti-infectives
Selectivity
The next-generation of precision antimicrobials should exhibit high selectivity towards pathogens and specificity against the desired targeted species. Synthetic peptides are promising candidates, and although still at an early stage, these agents have been rationally engineered by tuning their physicochemical and structural properties that are directly related to the primary sequence (Fig. 9). The modifications proposed turn these rationally designed AMPs into selective molecules for targeted killing of specific microbes.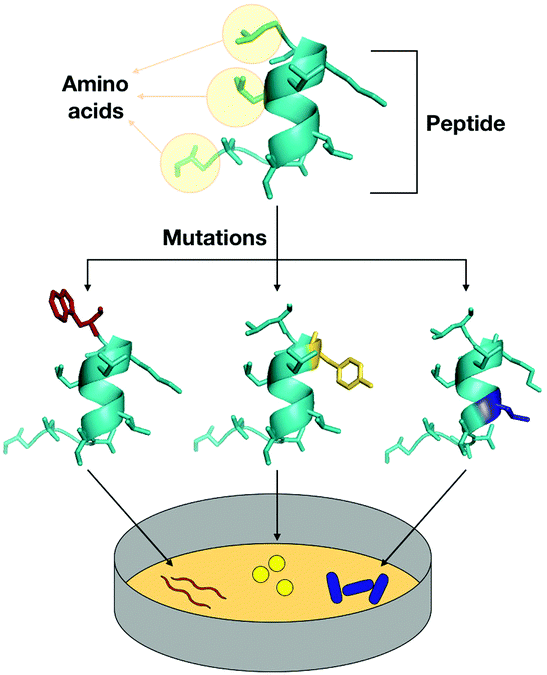 | ||
| Fig. 9 Synthetic designed peptides as targeted antimicrobial agents. | ||
A range of approaches have been developed to promote selectivity of targeted peptides, such as using intelligent-driven discovery algorithms based on information of non-cytotoxic peptides or evolving peptides considering properties that favor AMP-microorganism's membranes interactions. For instance, the algorithm Joker was used to rationally design the AMP PaDBS1R6, which presented selective antibacterial activities in vitro and in vivo against Gram-negative bacteria at concentrations as low as 16 μmol L−1.91 PaDBS1R6F10 (KKLRLKIAFK), which was designed by a sliding-window strategy on the basis of the 19-amino acid residue peptide, derived from a Pyrobaculum aerophilum ribosomal protein, exhibited anti-infective potential as it decreased the bacterial load in murine Pseudomonas aeruginosa cutaneous infections by more than 1000-fold. PaDBS1R6F10 did not exhibit cytotoxic and hemolytic effects against mammalian cells.
AMPs are mostly described to have activity against bacterial cells for the treatment or prevention of bacterial-related infections and disorders. However, AMPs can also be repurposed to target other types of cells such as pathogenic microorganisms (e.g., protozoa),103–110 or cancer cells.43,111 Decoralin, a broad-spectrum antibacterial AMP from the venom of the wasp Oreumenes decoratum, was recently reported to be inactive against Plasmodium species. However, by modifying the N-terminal extremity of this cytotoxic peptide, we were able to create synthetic derivatives having antiplasmodial activity at sub-micromolar concentrations that were not toxic against mammalian cells. Features such as positive net charge and hydrophobicity were fine-tuned in the N-terminal extremity, yielding the selective antiplasmodial agents.44 Decoralin was also repurposed into a selective anticancer agent through rational design, leading to the formation of necrotic pathways in breast cancer cells. Changes in hydrophobicity favoring the amphipathic structure, and helical conformations were described as being responsible for the anticancer activity of the decoralin analogs.43
Computational approaches for predicting active peptides
The diverse computational biology approaches available for drug discovery are a powerful tool for the accurate design of active peptides. The performance of methods varies depending on desired target and the availability of data and resources. The generation of more complex descriptors and scoring functions are key steps for developing more effective computationally aided drug design technologies.An alternative would be to couple methods based on structural data and those based in physicochemical features with molecular dynamics and molecular modeling, for example, the combination of quantum mechanics and molecular mechanics to create comprehensive suites to analyse target proteins or peptides and generate precise output data, such as the one described by Melo et al..112 The authors merged the two widely used molecular dynamics and visualization softwares NAMD and VMD with the quantum chemistry packages ORCA and MOPAC, demonstrating that interface, setup, execution, visualization, and analysis can be straightforward procedures for all levels of expertise.
Another example of precise AMP prediction are initiatives to find or build molecules that are selectively active against microorganisms. Vishnepolsky et al.113 recently described a predictive model of small linear AMPs that present antimicrobial activity against particular Gram-negative strains. The authors accurately distinguished active peptides with specific activity against E. coli ATCC 25922 and P. aeruginosa ATCC 27853 using a semi-supervised machine-learning approach coupled to a density-based clustering algorithm. Veltri et al.114 reported a method that constructs and selects complex descriptors of AMPs based on sequence patterns. This method might provide a summary of antibacterial activity at the sequence level by recognizing the antimicrobial activity of a peptide and predicting its target selectivity based on models of activity against Gram-positive and Gram-negative bacteria.
Self-assembled peptides
Self-assembled molecules, such as lipids, sugars, nucleic acids, proteins, and peptides are fundamental building blocks comprising cell membranes, cell cytoskeletal structures and extracellular matrices.115 Understanding peptide folding and self-assembling is still a challenge even with recent advances in computational biology, data-mining and experimental techniques.116 Due to their small size compared to proteins, peptides are dynamic molecules that tend to transition from disordered to helical, β- or γ-turns or stranded structures depending on the surrounding environment. AMPs are well-known for adopting defined structures in the presence of hydrophobic/hydrophilic interfaces, such as the interface of solvents and the membrane of microorganisms, because of their amphipathic sequence. Most of the known AMPs are helical when in contact with the membrane of cells. However, the composition of cells deeply influence the mechanism of action of peptides and their potency to destabilize the lipid bilayer that compose the biological membranes.28The self-assembly of peptides has been explored as a feature for understanding or modifying biological activities, besides preventing peptide exposure to enzymatic degradation.117 Self-assembled peptides have been used as building blocks for the generation of smart supramolecular nanomaterials used for biomedical applications, such as drug delivery, tissue engineering, antibacterial agents, and nanosensors.115,118 Some of the advantages of the self-assembled peptide nanostructures are chemical diversity, biocompatibility, high loading capacity for both hydrophobic and hydrophilic drugs, and their ability to respond to stimuli or to target molecular recognition sites or specific membrane compositions.118 However, understanding how peptides or small proteins fold and how to design amino acid portions so that they adopt certain geometrical arrangements is still the greatest obstacle to taking full advantage of self-assembled amphiphilic peptides. Several efforts have been made to tackle this issue, mostly using computational resources to predict ordering and tendency of each specific amino acid to adopt certain conformations during folding.119,120 The de novo design of self-assembled peptides and proteins is still the most widely accepted approach to the problem because of experimental limitation in obtaining dynamic properties during structuring.121 Thus, new ways to effectively combine computational and experimental methods are needed to increase our understanding of the specific role of the amino acid sequences in the supramolecular self-assembly of these molecules.
Merging synthetic biology, physicochemistry and computational biology approaches for the discovery, design and production of antimicrobial peptides
We envision that the next steps towards the translation of AMPs into clinical use will involve merging the discovery and design of novel selective and potent AMPs by means of computer science, automation and high-throughput chemical synthesis and screening. This would be followed by analysing the biological activity of these molecules and their production through synthetic biology, thus significantly reducing cost and labor (Fig. 10).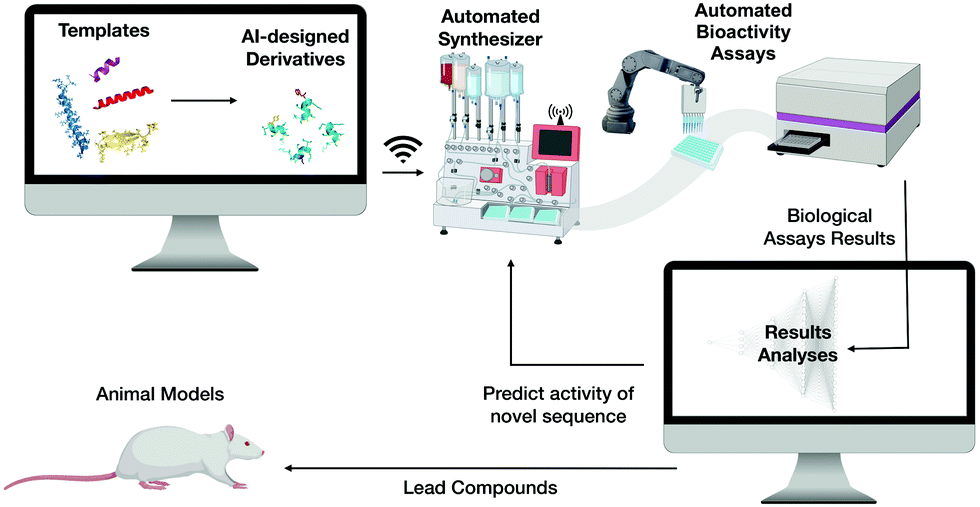 | ||
| Fig. 10 Autonomous platform for the discovery, design, synthesis, and biological screening of AMPs. | ||
Achieving this will enable the creation of a machine capable of autonomous molecular discovery and screening. The development of efficient technologies to automate discovery is the most promising of the remaining steps: several new approaches have already been described and new artificial intelligence platforms have been implemented12,24,29,31,32,40,91,92 and integrated with high-throughput systems.122
Other potential applications of reprogrammed peptides: peptide-based delivery-systems and materials
Their diversity and versatility have made peptides promising candidates not only for antimicrobial, antibiofilm, and immunomodulatory purposes, but also for additional multifunctional applications.28 Many classes of AMPs have been reported to be useful components of chemical/electrochemical biosensors for the detection of microorganisms,123,124 in which the affinity of the AMP for bacterial membranes is exploited to detect microorganisms through binding or peptide-membrane interactions. For example, a nanostructured biosensor has been described by Miranda et al. in which the AMP clavanin A (FLPIIVFQFLGKIIHHVGNFVHGFSHVF-NH2) was used for the detection of Gram-negative bacteria.125 The authors attributed the high sensitivity of the biosensing system to the specificity of clavanin A to Gram-negative bacteria. Another example is the design and use of a vancomycin derivative for the specific detection of Gram-positive bacteria on an impedance sensor. The sensor was based on the efficient capture of Gram-positive bacteria from the surrounding areas of the sensor by the vancomycin derivative molecules that were deposed as a thin film over the sensor surface.126 The rational design of peptides with higher affinity for specific bacterial strains or genera will lead to more sensitive and accurate detection and diagnostic systems.Peptide-based smart bioresponsive materials that are sensitive to biological or chemical changes in their environments are very well-known and explored platforms for the detection or delivery of other biomolecules.127 They are typically used for therapeutic purposes, such as diagnosis, drug delivery, tissue engineering and for constructing biomedical devices, but also for the generation of biodegradable materials and other types of responsive materials such as the multifunctional hydrogels. In comprehensive reviews Mart et al.128 and Lu et al.129 describe how engineered peptide-based materials can be constructed according to their properties and responsiveness.
Engineered peptides have been used for unusual applications, such as scaffolds for a templated chromophore assembly for artificial light-harvesting systems; here, modified peptides served to create tailored antenna architectures with yellow, red, and blue chromophores, exploiting three dynamic covalent reactions simultaneously, disulfide exchange, acyl hydrazone, and boronic ester formations.130 Additionally, peptides have been used for the generation of a supercapacitor with improved perfomance131 and piezoelectric materials for the creation of a flexible generator.132
Conclusions and outlook
The unique physicochemical and structural properties of engineered peptides render them a variety of three-dimensional scaffolds that, when coupled to computational biology tools, allow the extrapolation of peptides for the development of new biotechnological applications. In this Feature Article, we have described the importance of rational design, including biological, physicochemical, and structural descriptors, for the discovery and design of engineered, biologically active peptides. In particular, advances in the generation of biological activity data by high-throughput experimental platforms provide the necessary raw material for subsequent computer-based design and discovery. Successful methods to date include pattern recognition approaches and GAs. In turn, these techniques can be used to gain an understanding of how the structural and physicochemical properties of peptides influence their diverse biological activities. It has been reported that peptides can be reprogrammed to act selectively, an interesting biological property that may be incorporated to design novel antimicrobial, antibiofilm and immunomodulatory agents and to build peptides as carriers for drug delivery. The fine-tuned control of the properties of biologically active peptides remains a challenge, because the influence of several activity and structural descriptors on function is still unclear. We believe that structure–function-guided and deep learning approaches, coupled with dynamic simulation analyses of these molecules will help elucidate the role of the most important biological activity descriptors of peptides. Reprogramming peptides represents an exciting avenue for combatting infections caused by microorganisms and for generating forthcoming smart technologies, such as biosensors, stimuli-responsive materials, and drug delivery scaffolds.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Cesar de la Fuente-Nunez holds a Presidential Professorship at the University of Pennsylvania. The figures shown here were prepared using the Motifolio and Biorender drawing toolkits and PyMOL (v1.8.2.3).References
- C. de la Fuente-Nunez, M. D. Torres, F. J. Mojica and T. K. Lu, Curr. Opin. Microbiol., 2017, 37, 95–102 CrossRef CAS.
- V. Lázár, A. Martins, R. Spohn, L. Daruka, G. Grézal, G. Fekete, M. Számel, P. K. Jangir, B. Kintses, B. Csörgo, Á. Nyerges, Á. Györkei, A. Kincses, A. Dér, F. R. Walter, M. A. Deli, E. Urbán, Z. Hegedus, G. Olajos, O. Méhi, B. Bálint, I. Nagy, T. A. Martinek, B. Papp and C. Pál, Nat. Microbiol., 2018, 3, 718–731 CrossRef.
- K. L. Brown and R. E. W. Hancock, Curr. Opin. Immunol., 2006, 18, 24–30 CrossRef CAS.
- R. E. W. Hancock, Lancet Infect. Dis., 2001, 1, 156–164 CrossRef CAS.
- R. E. W. Hancock, Expert Opin. Invest. Drugs, 2000, 9, 1723–1729 CrossRef CAS PubMed.
- C. de la Fuente-Núñez, M. H. Cardoso, E. de Souza Cândido, O. L. Franco and R. E. W. Hancock, Biochim. Biophys. Acta, Biomembr., 2016, 1858, 1061–1069 CrossRef PubMed.
- D. Pletzer and R. E. W. Hancock, J. Bacteriol., 2016, 198, 2572–2578 CrossRef CAS.
- A. L. Hilchie, K. Wuerth and R. E. W. Hancock, Nat. Chem. Biol., 2013, 9, 761 CrossRef CAS.
- A. Nijnik and R. E. W. Hancock, Emerg. Health Threats J., 2009, 2, 7078 CrossRef.
- R. E. W. Hancock and H.-G. Sahl, Nat. Biotechnol., 2006, 24, 1551 CrossRef CAS.
- C. D. Fjell, J. A. Hiss, R. E. W. Hancock and G. Schneider, Nat. Rev. Drug Discovery, 2012, 11, 37–51 CrossRef CAS PubMed.
- W. F. Porto, A. S. Pires and O. L. Franco, Biotechnol. Adv., 2017, 35, 337–349 CrossRef CAS.
- A. Tossi, L. Sandri and A. Giangaspero, Biopolymers, 2000, 55, 4–30 CrossRef CAS PubMed.
- Z. Jiang, C. T. Mant, M. Vasil and R. S. Hodges, Chem. Biol. Drug Des., 2017, 91, 75–92 CrossRef.
- M. R. Yeaman and N. Y. Yount, Pharmacol. Rev., 2003, 55, 27–55 CrossRef CAS PubMed.
- L. T. Nguyen, E. F. Haney and H. J. Vogel, Trends Biotechnol., 2011, 29, 464–472 CrossRef CAS.
- T.-H. Lee, K. N. Hall and M.-I. Aguilar, Curr. Top. Med. Chem., 2016, 16, 25–39 CrossRef CAS PubMed.
- D. I. Andersson, D. Hughes and J. Z. Kubicek-Sutherland, Drug Resist. Updates, 2016, 26, 43–57 CrossRef CAS.
- M. Mahlapuu, J. Håkansson, L. Ringstad and C. Björn, Front. Cell. Infect. Microbiol., 2016, 6, 1–12 Search PubMed.
- R. J. Citorik, M. Mimee and T. K. Lu, Nat. Biotechnol., 2014, 32, 1141 CrossRef CAS.
- M. Mondhe, A. Chessher, S. Goh, L. Good and J. E. M. Stach, PLoS One, 2014, 9, e89082 CrossRef.
- H. Jenssen, Expert Opin. Drug Discovery, 2011, 6, 171–184 CrossRef CAS PubMed.
- R. Todeschini and V. Consonni, Molecular Descriptors for Chemoinformatics: Volume I: Alphabetical Listing/Volume II: Appendices, References, 2009 Search PubMed.
- K. Pane, L. Durante, O. Crescenzi, V. Cafaro, E. Pizzo, M. Varcamonti, A. Zanfardino, V. Izzo, A. Di Donato and E. Notomista, J. Theor. Biol., 2017, 419, 254–265 CrossRef CAS PubMed.
- K. Pane, V. Cafaro, A. Avitabile, M. D. T. Torres, A. Vollaro, E. De Gregorio, M. R. Catania, A. Di Maro, A. Bosso, G. Gallo, A. Zanfardino, M. Varcamonti, E. Pizzo, A. Di Donato, T. K. Lu, C. de la Fuente-Nunez and E. Notomista, ACS Synth. Biol., 2018, 7, 2105–2115 CrossRef CAS PubMed.
- K. Pane, L. Durante, E. Pizzo, M. Varcamonti, A. Zanfardino, V. Sgambati, A. Di Maro, A. Carpentieri, V. Izzo, A. Di Donato, V. Cafaro and E. Notomista, PLoS One, 2016, 11, e0146552 CrossRef PubMed.
- W. F. Porto, Á. S. Pires and O. L. Franco, J. Theor. Biol., 2017, 426, 96–103 CrossRef CAS PubMed.
- M. D. T. Torres, S. Sothiselvam, T. K. Lu and C. de la Fuente-Nunez, J. Mol. Biol., 2019, 431(18), 3547–3567 CrossRef CAS.
- W. F. Porto, L. Irazazabal, E. S. F. Alves, S. M. Ribeiro, C. O. Matos, Á. S. Pires, I. C. M. Fensterseifer, V. J. Miranda, E. F. Haney, V. Humblot, M. D. T. Torres, R. E. W. Hancock, L. M. Liao, A. Ladram, T. K. Lu, C. de la Fuente-Nunez and O. L. Franco, Nat. Commun., 2018, 9, 1490 CrossRef.
- C. M. Agbale, J. K. Sarfo, I. K. Galyuon, S. A. Juliano, G. G. O. Silva, D. F. Buccini, M. H. Cardoso, M. D. T. Torres, A. M. Angeles-Boza, C. de la Fuente-Nunez and O. L. Franco, Biochemistry, 2019, 58, 3802–3812 CrossRef CAS PubMed.
- C. de la Fuente-Nunez, mSystems, 2019, 4(3), e00151-19 CrossRef PubMed.
- M. D. T. Torres and C. de la Fuente-Nunez, Curr. Opin. Microbiol., 2019, 51, 30–38 CrossRef CAS PubMed.
- A. Supady, V. Blum and C. Baldauf, J. Chem. Inf. Model., 2015, 55, 2338–2348 CrossRef CAS PubMed.
- A. Lavecchia, Drug Discovery Today, 2015, 20, 318–331 CrossRef PubMed.
- A. N. Lima, E. A. Philot, G. H. G. Trossini, L. P. B. Scott, V. G. Maltarollo and K. M. Honorio, Expert Opin. Drug Discovery, 2016, 11, 225–239 CrossRef CAS PubMed.
- M. Yoshida, T. Hinkley, S. Tsuda, Y. M. Abul-Haija, R. T. McBurney, V. Kulikov, J. S. Mathieson, S. Galiñanes Reyes, M. D. Castro and L. Cronin, Chem, 2018, 4, 533–543 CAS.
- E. Y. Lee, B. M. Fulan, G. C. L. Wong and A. L. Ferguson, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 13588–13593 CrossRef CAS.
- Y. LeCun, Y. Bengio and G. Hinton, Nature, 2015, 521, 436 CrossRef CAS PubMed.
- A. T. Müller, J. A. Hiss and G. Schneider, J. Chem. Inf. Model., 2018, 58, 472–479 CrossRef.
- M. D. T. Torres, C. N. Pedron, Y. Higashikuni, R. M. Kramer, M. H. Cardoso, K. G. N. Oshiro, O. L. Franco, P. I. Silva Junior, F. D. Silva, V. X. Oliveira Junior, T. K. Lu and C. de la Fuente-Nunez, Commun. Biol., 2018, 1, 221 CrossRef PubMed.
- M. D. T. Torres, C. N. Pedron, J. A. da Silva Lima, P. I. da Silva, F. D. da Silva and V. X. Oliveira, J. Pept. Sci., 2017, 23, 818–823 CrossRef CAS.
- M. D. T. Torres, C. N. Pedron, I. Araújo, P. I. Silva, F. D. Silva and V. X. Oliveira, ChemistrySelect, 2017, 2, 18–23 CrossRef CAS.
- M. D. T. Torres, G. P. Andrade, R. H. Sato, C. N. Pedron, T. M. Manieri, G. Cerchiaro, A. O. Ribeiro, C. de la Fuente-Nunez and V. X. Oliveira, Beilstein J. Org. Chem., 2018, 14, 1693–1703 CrossRef CAS PubMed.
- M. D. T. Torres, A. F. Silva, C. N. Pedron, M. L. Capurro, C. de la Fuente-Nunez and V. X. O. Junior, ChemistrySelect, 2018, 3, 5859–5863 CrossRef CAS.
- E. F. Haney, S. C. Mansour, A. L. Hilchie, C. de la Fuente-Núñez and R. E. W. Hancock, Peptides, 2015, 71, 276–285 CrossRef CAS PubMed.
- K. Hilpert, R. Volkmer-Engert, T. Walter and R. E. W. Hancock, Nat. Biotechnol., 2005, 23, 1008 CrossRef CAS.
- N. Papo and Y. Shai, Biochemistry, 2004, 43, 6393–6403 CrossRef CAS.
- C. de la Fuente-Núñez, F. Reffuveille, S. C. Mansour, S. L. Reckseidler-Zenteno, D. Hernández, G. Brackman, T. Coenye and R. E. W. Hancock, Chem. Biol., 2015, 22, 196–205 CrossRef PubMed.
- T. Zhang, Z. Wang, R. E. W. Hancock, C. de la Fuente-Núñez and M. Haapasalo, PLoS One, 2016, 11, e0166997 CrossRef.
- M. Vert, Y. Doi, K.-H. Hellwich, M. Hess, P. Hodge, P. Kubisa, M. Rinaudo and F. Schué, Pure Appl. Chem., 2012, 84, 377–410 CAS.
- A. Di Grazia, F. Cappiello, H. Cohen, B. Casciaro, V. Luca, A. Pini, Y. P. Di, Y. Shai and M. L. Mangoni, Amino Acids, 2015, 47, 2505–2519 CrossRef CAS PubMed.
- M. Mardirossian, N. Pérébaskine, M. Benincasa, S. Gambato, S. Hofmann, P. Huter, C. Müller, K. Hilpert, C. A. Innis, A. Tossi and D. N. Wilson, Cell Chem. Biol., 2018, 25, 530–539.e7 CrossRef CAS PubMed.
- B. Deslouches, S. M. Phadke, V. Lazarevic, M. Cascio, K. Islam, R. C. Montelaro and T. A. Mietzner, Antimicrob. Agents Chemother., 2005, 49, 316–322 CrossRef CAS PubMed.
- R. W. Scott, W. F. DeGrado and G. N. Tew, Curr. Opin. Biotechnol., 2008, 19, 620–627 CrossRef CAS PubMed.
- C. H. Chen, C. G. Starr, E. Troendle, G. Wiedman, W. C. Wimley, J. P. Ulmschneider and M. B. Ulmschneider, J. Am. Chem. Soc., 2019, 141, 4839–4848 CrossRef CAS.
- R. Akbari, M. Hakemi Vala, A. Hashemi, H. Aghazadeh, J.-M. Sabatier and K. Pooshang Bagheri, Amino Acids, 2018, 50, 1231–1243 CrossRef CAS.
- A. T. Müller, G. Gabernet, J. A. Hiss and G. Schneider, Bioinformatics, 2017, 33, 2753–2755 CrossRef PubMed.
- M. Pillong, J. A. Hiss, P. Schneider, Y. C. Lin, G. Posselt, B. Pfeiffer, M. Blatter, A. T. Müller, S. Bachler, C. S. Neuhaus, P. S. Dittrich, K. H. Altmann, S. Wessler and G. Schneider, Small, 2017, 13, 1–11 Search PubMed.
- L. Tallorin, J. Wang, W. E. Kim, S. Sahu, N. M. Kosa, P. Yang, M. Thompson, M. K. Gilson, P. I. Frazier, M. D. Burkart and N. C. Gianneschi, Nat. Commun., 2018, 9, 5253 CrossRef.
- C. Beloin, S. Renard, J.-M. Ghigo and D. Lebeaux, Curr. Opin. Pharmacol., 2014, 18, 61–68 CrossRef CAS.
- C. de la Fuente-Núñez, S. C. Mansour, Z. Wang, L. Jiang, E. B. M. Breidenstein, M. Elliott, F. Reffuveille, D. P. Speert, S. L. Reckseidler-Zenteno, Y. Shen, M. Haapasalo and R. E. W. Hancock, Antibiotics, 2014, 3, 509–526 CrossRef.
- S. C. Mansour, C. de la Fuente-Núñez and R. E. W. Hancock, J. Pept. Sci., 2015, 21, 323–329 CrossRef CAS PubMed.
- S. M. Ribeiro, C. de la Fuente-Núñez, B. Baquir, C. Faria-Junior, O. L. Franco and R. E. W. Hancock, Antimicrob. Agents Chemother., 2015, 59, 3906–3912 CrossRef CAS PubMed.
- O. N. Silva, E. S. F. Alves, C. de la Fuente-Núñez, S. M. Ribeiro, S. M. Mandal, D. Gaspar, A. S. Veiga, M. A. R. B. Castanho, C. A. S. Andrade, J. M. Nascimento, I. C. M. Fensterseifer, W. F. Porto, J. R. Correa, R. E. W. Hancock, S. Korpole, A. L. Oliveira, L. M. Liao and O. L. Franco, Sci. Rep., 2016, 6, 27128 CrossRef CAS PubMed.
- C. de la Fuente-Núñez, F. Reffuveille, L. Fernández and R. E. W. Hancock, Curr. Opin. Microbiol., 2013, 16, 580–589 CrossRef PubMed.
- C. de la Fuente-Núñez, F. Reffuveille, E. F. Haney, S. K. Straus and R. E. W. Hancock, PLoS Pathog., 2014, 10, e1004152 CrossRef PubMed.
- E. Pizzo, K. Pane, A. Bosso, N. Landi, S. Ragucci, R. Russo, R. Gaglione, M. D. T. Torres, C. de la Fuente-Nunez, A. Arciello, A. Di Donato, E. Notomista and A. Di Maro, Biochim. Biophys. Acta, Biomembr., 2018, 1860, 1425–1435 CrossRef CAS PubMed.
- C. de la Fuente-Núñez, V. Korolik, M. Bains, U. Nguyen, E. B. M. Breidenstein, S. Horsman, S. Lewenza, L. Burrows and R. E. W. Hancock, Antimicrob. Agents Chemother., 2012, 56, 2696–2704 CrossRef PubMed.
- N. Bionda, R. M. Fleeman, C. de la Fuente-Núñez, M. C. Rodriguez, F. Reffuveille, L. N. Shaw, I. Pastar, S. C. Davis, R. E. W. Hancock and P. Cudic, Eur. J. Med. Chem., 2016, 108, 354–363 CrossRef CAS.
- F. Reffuveille, C. de la Fuente-Núñez, S. Mansour and R. E. W. Hancock, Antimicrob. Agents Chemother., 2014, 58, 5363–5371 CrossRef PubMed.
- T. Zhang, Z. Wang, R. E. W. Hancock, C. de la Fuente-Núñez and M. Haapasalo, PLoS One, 2016, 11, e0166997 CrossRef.
- C. de la Fuente-Núñez, V. Korolik, M. Bains, U. Nguyen, E. B. M. Breidenstein, S. Horsman, S. Lewenza, L. Burrows and R. E. W. Hancock, Antimicrob. Agents Chemother., 2012, 56, 2696–2704 CrossRef PubMed.
- M. P. Bevilacqua, D. J. Huang, B. D. Wall, S. J. Lane, C. K. Edwards, J. A. Hanson, D. Benitez, J. S. Solomkin and T. J. Deming, Macromol. Biosci., 2017, 17, 1600492 CrossRef PubMed.
- K. Hu, N. W. Schmidt, R. Zhu, Y. Jiang, G. H. Lai, G. Wei, E. F. Palermo, K. Kuroda, G. C. L. Wong and L. Yang, Macromolecules, 2013, 46, 1908–1915 CrossRef CAS.
- R. W. Scott and G. N. Tew, Curr. Top. Med. Chem., 2016, 17, 576–589 CrossRef PubMed.
- T. Stern, E. Zelinger and Z. Hayouka, Chem. Commun., 2016, 52, 7102–7105 RSC.
- A. Yehuda, L. Slamti, R. Bochnik-Tamir, E. Malach, D. Lereclus and Z. Hayouka, Chem. Commun., 2018, 54, 9777–9780 RSC.
- N. Bionda, R. M. Fleeman, C. de la Fuente-Núñez, M. C. Rodriguez, F. Reffuveille, L. N. Shaw, I. Pastar, S. C. Davis, R. E. W. Hancock and P. Cudic, Eur. J. Med. Chem., 2016, 108, 354–363 CrossRef CAS.
- C. de la Fuente-Núñez, O. N. Silva, T. K. Lu and O. L. Franco, Pharmacol. Ther., 2017, 178, 132–140 CrossRef PubMed.
- A. Nijnik and R. Hancock, Emerg. Health Threats J., 2009, 2, e1 CAS.
- E. F. Haney, S. C. Mansour, A. L. Hilchie, C. de la Fuente-Núñez and R. E. W. Hancock, Peptides, 2015, 71, 276–285 CrossRef CAS PubMed.
- C. G. Freitas, S. M. F. Lima, M. S. Freire, A. P. C. Cantuária, N. G. O. Júnior, T. S. Santos, J. S. Folha, S. M. Ribeiro, S. C. Dias, T. M. B. Rezende, P. Albuquerque, A. M. Nicola, C. de la Fuente-Núñez, R. E. W. Hancock, O. L. Franco and M. S. S. Felipe, Antimicrob. Agents Chemother., 2017, 61, e02518-16 CrossRef PubMed.
- O. N. Silva, C. de la Fuente-Núñez, E. F. Haney, I. C. M. Fensterseifer, S. M. Ribeiro, W. F. Porto, P. Brown, C. Faria-Junior, T. M. B. Rezende, S. E. Moreno, T. K. Lu, R. E. W. Hancock and O. L. Franco, Sci. Rep., 2016, 6, 35465 CrossRef CAS.
- L. H. J. Kleijn and N. I. Martin, in Antibacterials: Volume II, ed. J. F. Fisher, S. Mobashery and M. J. Miller, Springer International Publishing, Cham, 2018, pp. 27–53 Search PubMed.
- S. D. Taylor and M. Palmer, Bioorg. Med. Chem., 2016, 24, 6253–6268 CrossRef CAS PubMed.
- T. Tirilomis, Front. Immunol., 2014, 5, 97 Search PubMed.
- H. B. Koo and J. Seo, Pept. Sci., 2019, 111, e24122 Search PubMed.
- J. L. Lau and M. K. Dunn, Bioorg. Med. Chem., 2018, 26, 2700–2707 CrossRef CAS.
- L. M. Ballweber, J. E. Jaynes, W. E. Stamm and M. F. Lampe, Antimicrob. Agents Chemother., 2002, 46, 34–41 CrossRef CAS.
- M. Kudrimoti, A. Curtis, S. Azawi, F. Worden, S. Katz, D. Adkins, M. Bonomi, J. Elder, S. T. Sonis, R. Straube and O. Donini, J. Biotechnol., 2016, 239, 115–125 CrossRef CAS PubMed.
- I. C. M. Fensterseifer, M. R. Felício, E. S. F. Alves, M. H. Cardoso, M. D. T. Torres, C. O. Matos, O. N. Silva, T. K. Lu, M. V. Freire, N. C. Neves, S. Gonçalves, L. M. Lião, N. C. Santos, W. F. Porto, C. de la Fuente-Nunez and O. L. Franco, Biochim. Biophys. Acta, Biomembr., 2019, 1861, 1375–1387 CrossRef CAS PubMed.
- M. H. Cardoso, E. S. Cândido, L. Y. Chan, M. Der Torossian Torres, K. G. N. Oshiro, S. B. Rezende, W. F. Porto, T. K. Lu, C. de la Fuente-Nunez, D. J. Craik and O. L. Franco, ACS Infect. Dis., 2018, 4, 1727–1736 CrossRef CAS PubMed.
- P. R. Hansen and A. Oddo, in Fmoc Solid-Phase Peptide Synthesis, ed. G. Houen, Springer New York, New York, NY, 2015, pp. 33–50 Search PubMed.
- D. F. H. Winkler and K. Tian, Amino Acids, 2015, 47, 787–794 CrossRef CAS PubMed.
- R. Frank, J. Immunol. Methods, 2002, 267, 13–26 CrossRef CAS PubMed.
- H. Müller, D. Salzig and P. Czermak, Biotechnol. Prog., 2015, 31, 1–11 CrossRef.
- Y. Li, Protein Expression Purif., 2011, 80, 260–267 CrossRef CAS PubMed.
- T. Deng, H. Ge, H. He, Y. Liu, C. Zhai, L. Feng and L. Yi, Protein Expression Purif., 2017, 140, 52–59 CrossRef CAS PubMed.
- C. Schreiber, H. Müller, O. Birrenbach, M. Klein, D. Heerd, T. Weidner, D. Salzig and P. Czermak, Microb. Cell Fact., 2017, 16, 29 CrossRef PubMed.
- P. Perez-Pinera, N. Han, S. Cleto, J. Cao, O. Purcell, K. A. Shah, K. Lee, R. Ram and T. K. Lu, Nat. Commun., 2016, 7, 12211 CrossRef CAS PubMed.
- J. Cao, P. Perez-Pinera, K. Lowenhaupt, M.-R. Wu, O. Purcell, C. de la Fuente-Nunez and T. K. Lu, Nat. Commun., 2018, 9, 77 CrossRef PubMed.
- J. Cao, C. de la Fuente-Nunez, R. W. Ou, M. D. T. Torres, S. G. Pande, A. J. Sinskey and T. K. Lu, ACS Synth. Biol., 2018, 7, 896–902 CrossRef CAS PubMed.
- L. H. R. Ferreira, A. F. Silva, M. D. T. Torres, C. N. Pedron, M. L. Capurro, F. L. Alves, A. Miranda and V. X. Oliveira, Int. J. Pept. Res. Ther., 2014, 20, 553–564 CrossRef CAS.
- A. F. Silva, E. L. Bastos, M. D. T. Torres, A. L. Costa-Da-Silva, R. S. Ioshino, M. L. Capurro, F. L. Alves, A. Miranda, R. De Freitas Fischer Vieira and V. X. Oliveira, J. Pept. Sci., 2014, 20, 640–648 CrossRef CAS.
- M. Der Torossian Torres, A. F. Silva, F. L. Alves, M. L. Capurro, A. Miranda and V. X. Oliveira Jr., Int. J. Pept. Res. Ther., 2014, 20(3), 277–287 CrossRef CAS.
- T. Marcelo Der Torossian, A. F. Silva, F. L. Alves, M. L. Capurro, A. Miranda and O. Vani Xavier, Chem. Biol. Drug Des., 2015, 85, 163–171 CrossRef PubMed.
- M. D. T. Torres, A. F. Silva, F. L. Alves, M. L. Capurro, A. Miranda, R. M. Cordeiro and V. X. Oliveira, J. Pept. Sci., 2016, 22, 132–142 CrossRef CAS.
- A. F. Silva, F. L. Alves, C. N. Pedron, M. D. T. Torres, L. S. Silva, A. A. S. Pinheiro, A. Miranda and V. X. Oliveira, Bioorg. Med. Chem. Lett., 2015, 25, 3311–3313 CrossRef CAS PubMed.
- M. D. T. Torres, A. F. Silva, F. L. Alves, M. L. Capurro, A. Miranda, R. M. Cordeiro and V. X. Oliveira Junior, J. Pept. Sci., 2016, 22, 132–142 CrossRef CAS.
- M. Der Torossian Torres, A. F. Silva, L. De Souza Silva, A. A. De Sá Pinheiro and V. X. Oliveira, J. Pept. Sci., 2015, 21, 24–28 CrossRef PubMed.
- C. N. Pedron, G. P. Andrade, R. H. Sato, M. D. T. Torres, G. Cerchiaro, A. O. Ribeiro and V. X. Oliveira Jr., Chem. Biol. Drug Des., 2018, 91, 588–596 CrossRef CAS PubMed.
- M. C. R. Melo, R. C. Bernardi, T. Rudack, M. Scheurer, C. Riplinger, J. C. Phillips, J. D. C. Maia, G. B. Rocha, J. V. Ribeiro, J. E. Stone, F. Neese, K. Schulten and Z. Luthey-Schulten, Nat. Methods, 2018, 15, 351 CrossRef CAS.
- B. Vishnepolsky, A. Gabrielian, A. Rosenthal, D. E. Hurt, M. Tartakovsky, G. Managadze, M. Grigolava, G. I. Makhatadze and M. Pirtskhalava, J. Chem. Inf. Model., 2018, 58, 1141–1151 CrossRef CAS PubMed.
- D. Veltri, U. Kamath and A. Shehu, IEEE/ACM Trans. Comput. Biol. Bioinf., 2017, 14, 300–313 Search PubMed.
- L. Sun, C. Zheng and T. Webster, Int. J. Nanomed., 2016, 12, 73–86 CrossRef.
- W. Hu, B. T. Walters, Z.-Y. Kan, L. Mayne, L. E. Rosen, S. Marqusee and S. W. Englander, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 7684–7689 CrossRef CAS.
- G. Bhardwaj, V. K. Mulligan, C. D. Bahl, J. M. Gilmore, P. J. Harvey, O. Cheneval, G. W. Buchko, S. V. S. R. K. Pulavarti, Q. Kaas, A. Eletsky, P.-S. Huang, W. A. Johnsen, P. J. Greisen, G. J. Rocklin, Y. Song, T. W. Linsky, A. Watkins, S. A. Rettie, X. Xu, L. P. Carter, R. Bonneau, J. M. Olson, E. Coutsias, C. E. Correnti, T. Szyperski, D. J. Craik and D. Baker, Nature, 2016, 538, 329–335 CrossRef CAS PubMed.
- N. Habibi, N. Kamaly, A. Memic and H. Shafiee, Nano Today, 2016, 11, 41–60 CrossRef CAS PubMed.
- P. Hosseinzadeh, G. Bhardwaj, V. K. Mulligan, M. D. Shortridge, T. W. Craven, F. Pardo-Avila, S. A. Rettie, D. E. Kim, D.-A. Silva, Y. M. Ibrahim, I. K. Webb, J. R. Cort, J. N. Adkins, G. Varani and D. Baker, Science, 2017, 358, 1461–1466 CrossRef CAS PubMed.
- Z. Chen, M. C. Johnson, J. Chen, M. J. Bick, S. E. Boyken, B. Lin, J. J. De Yoreo, J. M. Kollman, D. Baker and F. DiMaio, J. Am. Chem. Soc., 2019, 141, 8891–8895 CrossRef CAS PubMed.
- S. W. Englander and L. Mayne, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 8253–8258 CrossRef CAS PubMed.
- A. J. Mijalis, D. A. Thomas, M. D. Simon, A. Adamo, R. Beaumont, K. F. Jensen and B. L. Pentelute, Nat. Chem. Biol., 2017, 13, 464–466 CrossRef CAS PubMed.
- N. V. Kulagina, M. E. Lassman, F. S. Ligler and C. R. Taitt, Anal. Chem., 2005, 77, 6504–6508 CrossRef CAS PubMed.
- M. Hoyos-Nogués, S. Brosel-Oliu, N. Abramova, F.-X. Muñoz, A. Bratov, C. Mas-Moruno and F.-J. Gil, Biosens. Bioelectron., 2016, 86, 377–385 CrossRef PubMed.
- J. L. de Miranda, M. D. L. Oliveira, I. S. Oliveira, I. A. M. Frias, O. L. Franco and C. A. S. Andrade, Biochem. Eng. J., 2017, 124, 108–114 CrossRef CAS.
- S. Singh, A. Moudgil, N. Mishra, S. Das and P. Mishra, Biosens. Bioelectron., 2019, 136, 23–30 CrossRef CAS PubMed.
- Y. Shen, X. Fu, W. Fu and Z. Li, Chem. Soc. Rev., 2015, 44, 612–622 RSC.
- R. J. Mart, R. D. Osborne, M. M. Stevens and R. V. Ulijn, Soft Matter, 2006, 2, 822 RSC.
- Y. Lu, A. A. Aimetti, R. Langer and Z. Gu, Nat. Rev. Mater., 2017, 2, 16075 CrossRef CAS.
- L. Rocard, D. Wragg, S. A. Jobbins, L. Luciani, J. Wouters, S. Leoni and D. Bonifazi, Chem. – Eur. J., 2018, 24, 16136–16148 CrossRef CAS PubMed.
- K. Hu, C. Zheng, M. An, X. Ma and L. Wang, J. Mater. Chem. A, 2018, 6, 8047–8052 RSC.
- K. Jenkins, S. Kelly, V. Nguyen, Y. Wu and R. Yang, Nano Energy, 2018, 51, 317–323 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2019 |