
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The hydrophobically-tagged MDM2–p53 interaction inhibitor Nutlin-3a-HT is more potent against tumor cells than Nutlin-3a†
Florian
Nietzold‡
,
Stefan
Rubner‡
and
Thorsten
Berg
 *
*
Institute of Organic Chemistry, Leipzig University, Johannisallee 29, 04103 Leipzig, Germany. E-mail: tberg@uni-leipzig.de; Fax: +49 341 9736599
First published on 5th November 2019
Abstract
We present the first application of hydrophobic tagging to a non-covalent inhibitor of protein–protein interactions. Nutlin-3a-HT, created by fusing the hydrophobic tag HyT13 to the MDM2–p53 interaction inhibitor Nutlin-3a, prevented cellular accumulation of MDM2 upon p53 reactivation, and had a stronger effect on cell viability and the induction of apoptosis than Nutlin-3a.
Loss of function of the tumor suppressor p53 by mutation or deletion is found in a large proportion of human tumors.1 In tumor cells bearing wild-type p53, the major mechanism of p53 inhibition is via overexpression of the E3 ubiquitin ligase MDM2, which inactivates p53 by binding to its transactivation domain, promoting nuclear export, and inducing its proteasomal degradation.2 Overexpression of MDM2 can be caused by gene amplification, which is found in 7% of human cancers, with the highest frequency of gene amplification observed in soft tissue tumors (20%).3 Inhibitors of the MDM2–p53 interaction relieve inhibition of p53 by binding to MDM2, leading to increased p53 concentrations and the induction of apoptosis in tumor cells.4 The Nutlin series of cis-imidazolines, including Nutlin-3a (1, Fig. 1A), bind to MDM2 in a reversible manner, and were the first potent small-molecule modulators of the MDM2–p53 interaction.5 Currently, a number of MDM2–p53 interaction inhibitors are being tested in clinical trials.6
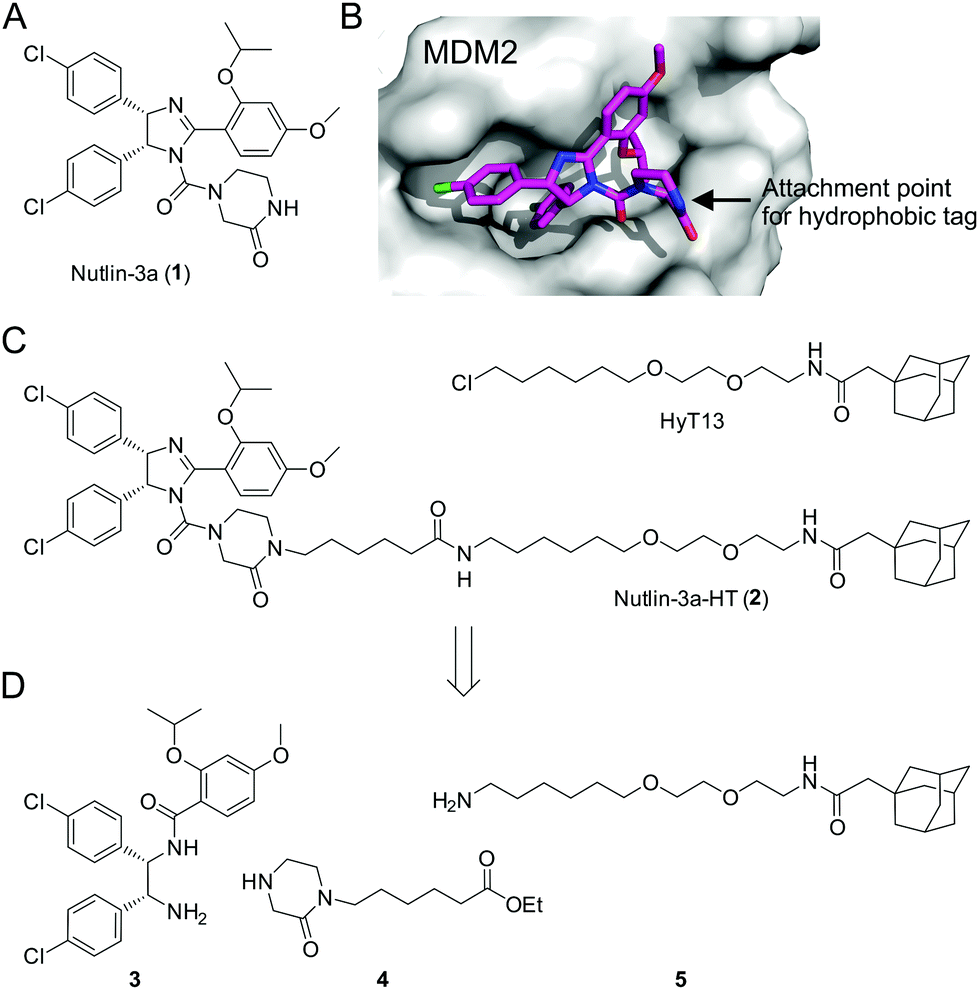 | ||
Fig. 1 (A) Structure of Nutlin-3a (1). (B) Co-crystal structure of 1 bound to MDM2 (PDB ID: 4J3E).14 (C) Structures of HyT13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 7a and Nutlin-3a-HT (2). (D) Building blocks for the synthesis of 2. 7a and Nutlin-3a-HT (2). (D) Building blocks for the synthesis of 2. | ||
Hydrophobic tagging of small molecules and peptides has been shown to be a powerful approach by which to degrade target proteins. Upon binding of a bioactive molecule to its binding partner, the hydrophobic tag is presented on the protein surface, mimicking a partially unfolded protein.7 This is thought to activate the cell's quality control machinery, and after a failed refolding attempt by chaperones, the protein is degraded by the proteasome.8 Applications of this concept include the pseudokinase Her3,9 the androgen receptor,10 dihydrofolate reductase,7b the E3 ligase cereblon,11 the DNA binding protein TDP-43,12 and the tau protein.13
We recently presented the first application of hydrophobic tagging to an existing inhibitor of a protein–protein interaction domain by fusing Poloxin-2,15 an irreversible inhibitor of the protein–protein interaction domain of the serine/threonine kinase Plk1, with two different hydrophobic adamantane-based tags,16 which led to proteasome-dependent degradation of the target protein.9a,10,16a In theory, irreversibly-binding bioactive molecules are preferable for the hydrophobic tagging approach, because their use results in a maximal display time of the hydrophobic tag on the protein surface. However, the vast majority of bioactive molecules designed in chemical biology and medicinal chemistry bind non-covalently to their protein targets. We aimed to explore the feasibility of expanding the application of hydrophobic tagging to include reversibly binding small-molecule inhibitors of protein–protein interactions.
The mode of binding of Nutlin-3a to MDM2 has been determined by X-ray crystallography.14 The two chlorophenyl moieties and the isopropoxy group of Nutlin-3a insert deeply into the hydrophobic pocket of MDM2 (Fig. 1B). In contrast, the piperazinone moiety is located at the rim of the hydrophobic pocket, with the amide nitrogen pointing towards the solvent. On the basis of this structure, we designed the fusion molecule 2 dubbed Nutlin-3a-HT, in which a hydrophobic tag based on HyT137a is connected to the piperazinone moiety of 1via the amide nitrogen (Fig. 1C). Binding of 2 to MDM2 should result in the presentation of the hydrophobic tag on the surface of MDM2, without significantly interfering with protein binding.
Synthesis of 2 was achieved from the imidazoline precursor 3, the piperazinone 4, and the hydrophobic tag amine 5 (Fig. 1D). Compound 3 was obtained in a variation of the protocol for Nutlin-3a synthesis developed by Johnston and co-workers17 in diastereomerically pure form and an enantiomeric excess (e.e.) of 99% (Fig. S1–S3, ESI†). The piperazinone building block 4 was synthesized in a 4-step procedure from piperazin-2-one by N-Boc protection, N-alkylation with 6-bromohexanoic acid, esterification and N-Boc deprotection in 53% yield (Fig. 2A). N-Alkylation of N-Boc-protected piperazin-2-one with bromohexanoic acid ethyl ester was not successful. The hydrophobic amine 5 is generated via Staudinger reaction from the azide 6 (Fig. 2B), which itself is accessible in 5 synthetic steps.16N-Acetylation of 5 afforded the control compound 7, which lacks the MDM2-recognition elements of 2.
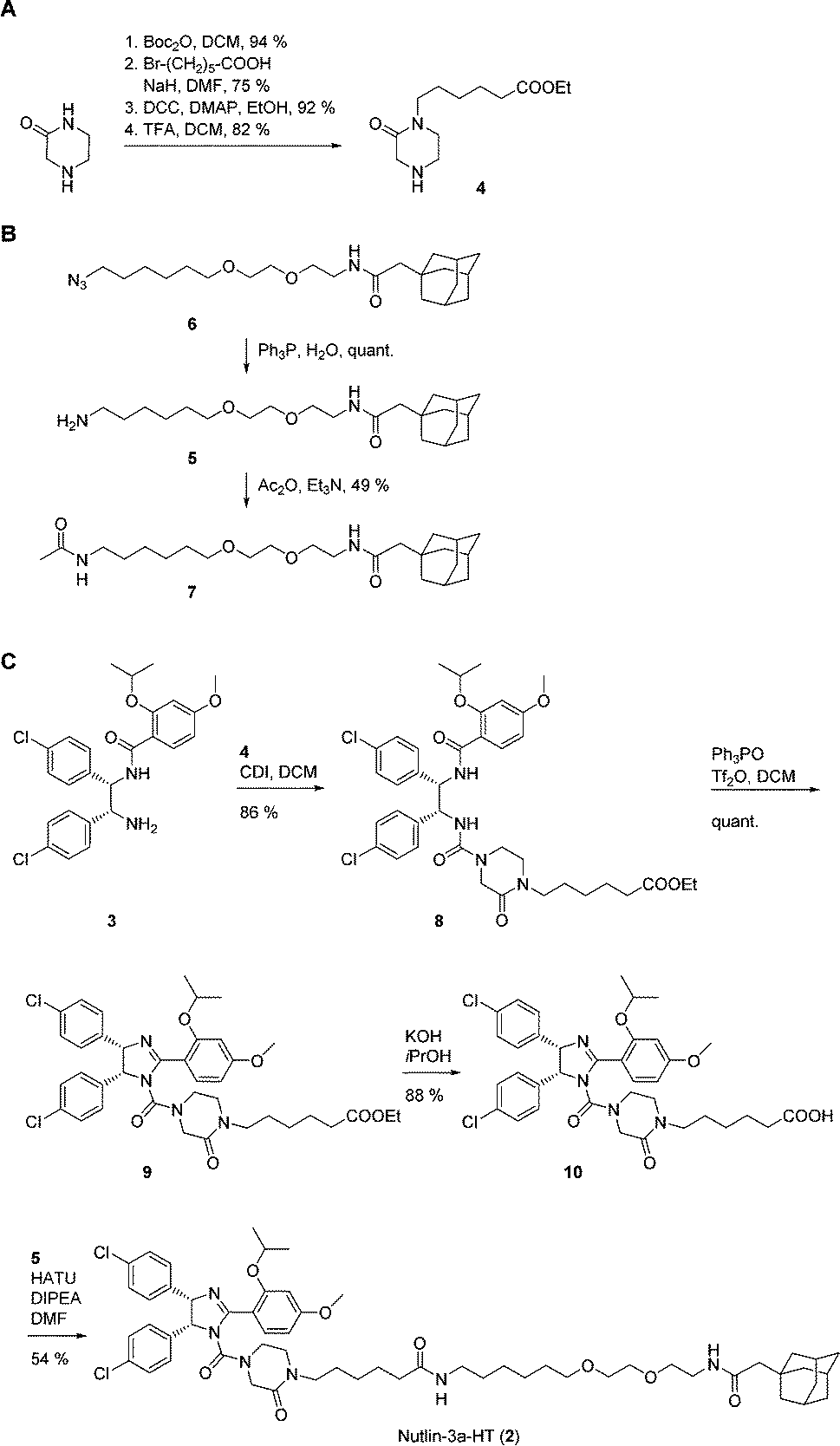 | ||
| Fig. 2 Synthesis of (A) piperazinone 4, (B) hydrophobic tag amine 5 and control compound 7, and (C) Nutlin-3a-HT (2). | ||
Coupling of the piperazinone 4 to 3 afforded the diamide 8, which was cyclized to the cis-imidazoline 9 using the Hendrickson reagent18 (Fig. 2C). Ester hydrolysis to 10 and coupling to the hydrophobic tag amine 5 provided Nutlin-3a-HT (2) in a procedure that involved a total of 23 synthetic steps based on commercially available starting materials.
In a fluorescence polarization (FP)-based assay, Nutlin-3a-HT (2) displaced a fluorophore-labeled, p53-derived peptide from MDM2 with low micromolar activity (IC50 = 2.83 ± 0.40 μM, Ki = 1.24 ± 0.18 μM)19 (Fig. 3). Although this represents an 8-fold lower activity against MDM2 compared to Nutlin-3a (1) (IC50 = 0.35 ± 0.03 μM, Ki = 0.15 ± 0.01 μM),19 previous studies have also observed a modest loss in activity upon conjugation of a bioactive molecule to a hydrophobic tag.10,16b Compound 7 did not inhibit the MDM2–p53 interaction, indicating its suitability for use as negative control compound in cell-based assays.
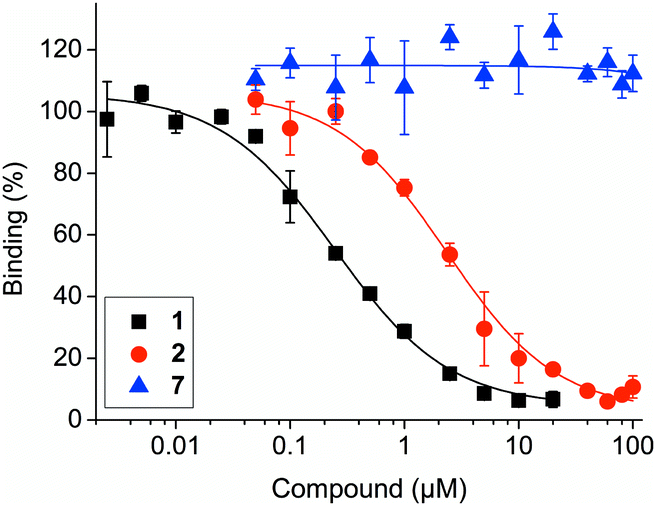 | ||
| Fig. 3 Activities of Nutlin-3a (1), Nutlin-3a-HT (2) and control compound 7 against the MDM2–p53 interaction in competitive fluorescence polarization assays. | ||
Exposure of HCT-116 (human colon carcinoma) cells, which carry wild-type p53, to Nutlin-3a (1) for 24 h induced a dose-dependent increase in the protein levels of MDM2, p53, and the cell cycle inhibitor p21 (Fig. 4A). These observations are consistent with the literature and are expected for a MDM2–p53 interaction inhibitor, since both MDM2 and p21 are transcriptionally activated by p53. Nevertheless, the observation that the levels of MDM2 are increased to a higher extent than the levels of p53 and p21 (Fig. 4B) by 1 represents an inherent drawback of occupancy-based MDM2–p53 interaction inhibitors, which is likely to limit their clinical efficacy.
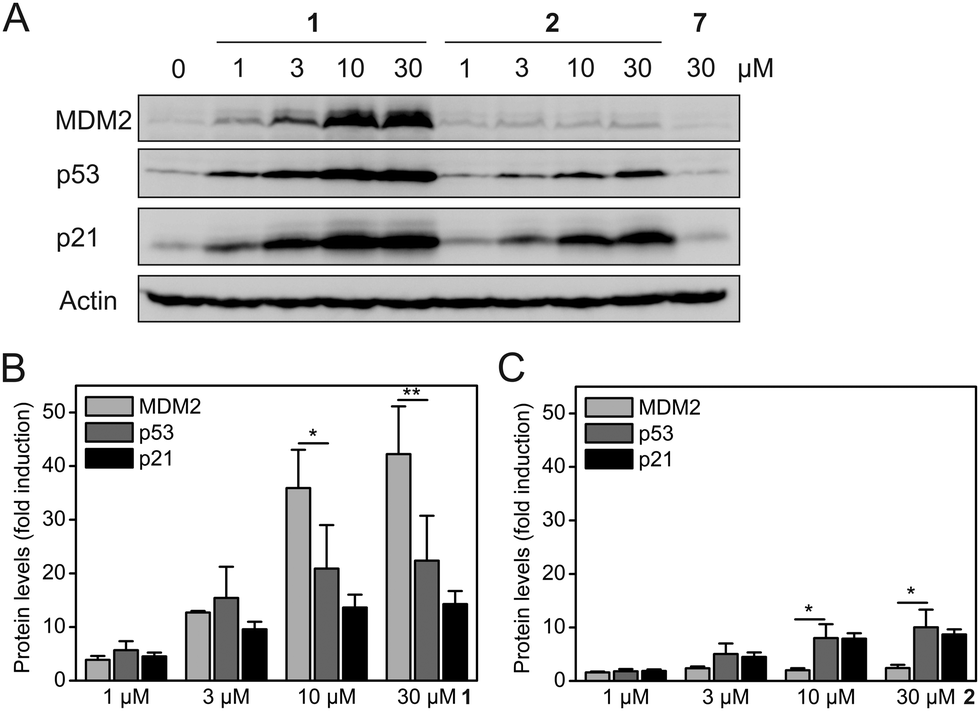 | ||
| Fig. 4 (A) Effect of Nutlin-3a (1), Nutlin-3a-HT (2), and control compound 7 on the protein levels of MDM2, p53, and p21 (n = 3). (B and C) Fold increase of the protein levels of MDM2, p53, and p21 compared to the untreated DMSO control in the presence of (B) compound 1 and (C) compound 2. Mean values and standard deviations are shown. *p < 0.05, **p < 0.01 (t-test, two-tailed, paired). | ||
In contrast, exposure of HCT-116 cells to hydrophobically-tagged Nutlin-3a-HT (2) for 24 h resulted in only a minimal increase of MDM2 levels, whilst inducing a much stronger and dose-dependent increase in the levels of p53 and its transcriptional target p21 (Fig. 4A and Fig. S4, ESI†). The concentrations of p53 and p21 in cells treated with 2 are raised to a higher extent than the concentrations of MDM2 (Fig. 4C). This suggests that the degradation-inducing mechanism of action of 2 is superior to the occupancy-based mechanism of action of 1. The control compound 7 did not alter the protein levels of any of the three proteins (Fig. 4A), excluding any non-specific effect of the adamantyl group.
Release of MDM2-induced p53 inhibition by treatment with the untagged Nutlin-3a (1) leads to a significant increase in the levels of both MDM2 and p21 (Fig. 4 and Fig. S4, ESI†), with 30 μM 1 inducing a 42-fold increase in MDM2 and a 14-fold increase in p21. In contrast, 30 μM of the hydrophobically-tagged Nutlin-3a-HT (2) increases p21 levels significantly more strongly than MDM2 levels (8.7-fold versus 2.4-fold) (Fig. 4 and Fig. S4, ESI†). This observation, in combination with the known degradation-targeting properties of the hydrophobic tag HyT13,7a,16a supports the idea that binding and degradation of MDM2 is the mechanism of action of 2. The much weaker induction of MDM2 by 2 as compared to 1 cannot be attributed to the reduced affinity for MDM2 seen in FP assays (Fig. 3), because this would not explain the increased p53 and p21 levels in the presence of 2 (Fig. 4A).
We note that the levels of p53 in cells treated with 2 are generally lower than in cells treated with 1 (Fig. 4A). We hypothesize that proteasomal degradation of MDM2 by 2 may not be limited to MDM2 itself, but may also affect, to a lesser extent, proteins bound to the MDM2 molecules marked for proteasomal degradation. Consistent with this model, proteasomal degradation of a non-ubiquitinated protein when bound to an ubiquitinated protein has been reported.20 It is conceivable that an exchange of 2 for p53 as the binding partner of MDM2 may occur in the window after the degradation of a particular MDM2 molecule has been decided by the cell's quality control machinery, leading to concomitant degradation of p53. This model would also explain the slightly reduced protein levels of the p53 transcriptional target p21 in the presence of 2 (Fig. 4A). In a recent study on MDM2-targeting PROTACs based on the MDM2–p53 interaction inhibitor MI-1061 and cereblon ligands, a similar effect was observed for some of the PROTACS.21
Consistent with the results of the Western blots, HCT-116 cells treated with 2 showed a higher degree of apoptosis than cells treated with 1 did (39 ± 7% apoptotic cells for 30 μM 2vs. 15 ± 1% for 30 μM 1, Fig. 5A and Fig. S5, ESI†). The Western blot data (Fig. 4) and the apoptosis assay data (Fig. 5A) are also consistent with the results of cell viability assays obtained after the same time of compound exposure (24 h), which show a dose-dependent reduction of cell viability for either compound, with stronger effects observed for 2 (Fig. 5B). The efficacy of both 1 and 2 increases with longer exposure times of 48 h and 120 h. After 48 h of exposure, 2 had an IC50 value of 10.5 ± 2.5 μM, whereas 1 only exhibited a 41% reduction of cell viability at 30 μM (Fig. S6, ESI†). After 120 h, 2 exhibited an IC50 value of 0.83 ± 0.17 μM, which is 2.4-fold lower compared to 1 (IC50 = 2.00 ± 0.09 μM, Fig. 5C).
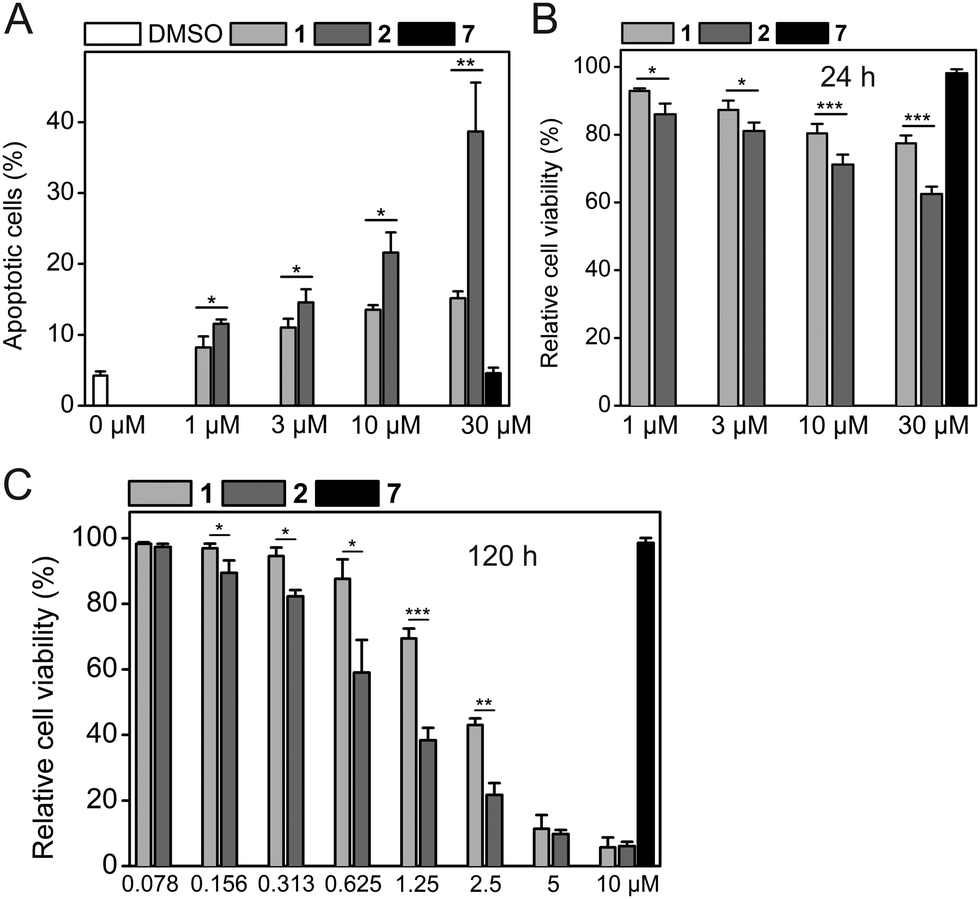 | ||
| Fig. 5 (A) Rate of apoptosis in HeLa cells treated with 1, 2, and control compound 7 for 24 h as analyzed by flow cytometry (n = 4). (B and C) Cell viability assays of HCT-116 cells after (B) 24 h, and (C) 120 h exposure to 1, 2, and 7 (n = 3). Cell viability in the presence of DMSO control was defined as 100%. Mean values and standard deviations are shown. *p < 0.05, **p < 0.01, ***p < 0.001 (t-test, two-tailed, paired). | ||
Because of the high activity of both 1 and 2 after 120 h, both compounds had a similar effect on cell viability at the highest concentrations tested (5 μM and 10 μM). The negative control compound 7 did not affect the apoptotic rate or cell viability of HCT-116 cells under any of the conditions tested, excluding non-specific effects caused by the hydrophobic tag as the reason for the superior activity of 2. Together with the known MDM2-selective effects of 1,5 combined cell-based data are consistent with selective binding and degradation of MDM2 as the mechanism of action of Nutlin-3a-HT (2).
In this study, we report the synthesis and evaluation of Nutlin-3a-HT (2) as the first hydrophobically-tagged small-molecule inhibitor of the MDM2–p53 interaction. Cell-based data for 2 indicate that it targets MDM2 for degradation in human tumor cells. 2 had a stronger effect on cell viability and the induction of apoptosis than the untagged MDM2–p53 interaction inhibitor Nutlin-3a (1). While the degrading potency of 2 is lower than that of some MDM2-targeting PROTACS,21 a major advantage of the hydrophobic tagging approach over the PROTAC approach using cereblon ligands is the guaranteed absence of teratogenicity mediated by the cereblon ligand, either in the context of the intact PROTAC or via metabolites.16a In addition, the hydrophobic tagging approach has the potential to be less dependent on the chemical nature and length of the linker connecting the protein-binding entity and the moiety facilitating protein degradation than the PROTAC-based approach is. Our data demonstrate that hydrophobic tagging, a method that was previously shown to improve the cellular potency of an existing irreversible inhibitor of a protein–protein interaction domain,16 can also be applied to reversibly binding small-molecule inhibitors of protein–protein interaction domains. Future studies will be required to define the requirements for potent hydrophobically-tagged, reversible inhibitors of protein–protein interactions in terms of activities and/or dissociation rates. Our work vastly expands the scope of hydrophobic tagging of pre-existing small-molecule inhibitors of protein–protein interactions as a method by which to target and degrade disease-related proteins.
This work was generously supported by the Deutsche Forschungsgemeinschaft (BE 4572/3-1), as well as the European Union and the Free State of Saxony, European Regional Development Fund. We thank the Core Unit Fluorescence Technologies of the Interdisciplinary Centre for Clinical Research (IZKF) at the Faculty of Medicine of Leipzig University (Kathrin Jäger and Andreas Lösche) for support with the flow cytometry analysis, Barbara Klüver for experimental support, and Angela Berg for critical reading of the manuscript.
Conflicts of interest
There are no conflicts of interest to declare.Notes and references
- D. Hamroun, S. Kato, C. Ishioka, M. Claustres, C. Beroud and T. Soussi, Hum. Mutat., 2006, 27, 14–20 CrossRef CAS PubMed.
- D. A. Freedman, L. Wu and A. J. Levine, Cell. Mol. Life Sci., 1999, 55, 96–107 CrossRef CAS PubMed.
- J. Momand, D. Jung, S. Wilczynski and J. Niland, Nucleic Acids Res., 1998, 26, 3453–3459 CrossRef CAS PubMed.
- N. Estrada-Ortiz, C. G. Neochoritis and A. Dömling, ChemMedChem, 2016, 11, 757–772 CrossRef CAS PubMed.
- L. T. Vassilev, B. T. Vu, B. Graves, D. Carvajal, F. Podlaski, Z. Filipovic, N. Kong, U. Kammlott, C. Lukacs, C. Klein, N. Fotouhi and E. A. Liu, Science, 2004, 303, 844–848 CrossRef CAS PubMed.
- S. Wang, Y. Zhao, A. Aguilar, D. Bernard and C. Y. Yang, Cold Spring Harbor Perspect. Med., 2017, 7, a026245 CrossRef PubMed.
- (a) T. K. Neklesa, H. S. Tae, A. R. Schneekloth, M. J. Stulberg, T. W. Corson, T. B. Sundberg, K. Raina, S. A. Holley and C. M. Crews, Nat. Chem. Biol., 2011, 7, 538–543 CrossRef CAS PubMed; (b) M. J. Long, D. R. Gollapalli and L. Hedstrom, Chem. Biol., 2012, 19, 629–637 CrossRef CAS PubMed.
- (a) T. K. Neklesa and C. M. Crews, Nature, 2012, 487, 308–309 CrossRef CAS PubMed; (b) P. M. Cromm and C. M. Crews, Cell Chem. Biol., 2017, 24, 1181–1190 CrossRef CAS PubMed.
- (a) T. Xie, S. M. Lim, K. D. Westover, M. E. Dodge, D. Ercan, S. B. Ficarro, D. Udayakumar, D. Gurbani, H. S. Tae, S. M. Riddle, T. Sim, J. A. Marto, P. A. Janne, C. M. Crews and N. S. Gray, Nat. Chem. Biol., 2014, 10, 1006–1012 CrossRef CAS PubMed; (b) S. M. Lim, T. Xie, K. D. Westover, S. B. Ficarro, H. S. Tae, D. Gurbani, T. Sim, J. A. Marto, P. A. Janne, C. M. Crews and N. S. Gray, Bioorg. Med. Chem. Lett., 2015, 25, 3382–3389 CrossRef CAS PubMed.
- J. L. Gustafson, T. K. Neklesa, C. S. Cox, A. G. Roth, D. L. Buckley, H. S. Tae, T. B. Sundberg, D. B. Stagg, J. Hines, D. P. McDonnell, J. D. Norris and C. M. Crews, Angew. Chem., Int. Ed., 2015, 54, 9659–9662 CrossRef CAS PubMed.
- C. Steinebach, I. Sosic, S. Lindner, A. Bricelj, F. Kohl, Y. L. D. Ng, M. Monschke, K. G. Wagner, J. Kronke and M. Gütschow, MedChemComm, 2019, 10, 1037–1041 RSC.
- N. Gao, Y. P. Huang, T. T. Chu, Q. Q. Li, B. Zhou, Y. X. Chen, Y. F. Zhao and Y. M. Li, Bioorg. Chem., 2019, 84, 254–259 CrossRef CAS PubMed.
- N. Gao, T.-T. Chu, Q.-Q. Li, Y.-J. Lim, T. Qiu, M.-R. Ma, Z.-W. Hu, X.-F. Yang, Y.-X. Chen, Y.-F. Zhao and Y.-M. Li, RSC Adv., 2017, 7, 40362–40366 RSC.
- B. Vu, P. Wovkulich, G. Pizzolato, A. Lovey, Q. Ding, N. Jiang, J. J. Liu, C. Zhao, K. Glenn, Y. Wen, C. Tovar, K. Packman, L. Vassilev and B. Graves, ACS Med. Chem. Lett., 2013, 4, 466–469 CrossRef CAS PubMed.
- A. Scharow, M. Raab, K. Saxena, S. Sreeramulu, D. Kudlinzki, S. Gande, C. Dotsch, E. Kurunci-Csacsko, S. Klaeger, B. Kuster, H. Schwalbe, K. Strebhardt and T. Berg, ACS Chem. Biol., 2015, 10, 2570–2579 CrossRef CAS PubMed.
- (a) S. Rubner, A. Scharow, S. Schubert and T. Berg, Angew. Chem., Int. Ed., 2018, 57, 17043–17047 CrossRef CAS PubMed; (b) S. Rubner, S. Schubert and T. Berg, Org. Biomol. Chem., 2019, 17, 3113–3117 RSC.
- T. A. Davis, A. E. Vilgelm, A. Richmond and J. N. Johnston, J. Org. Chem., 2013, 78, 10605–10616 CrossRef CAS PubMed.
- (a) J. B. Hendrickson and M. S. Hussoin, J. Org. Chem., 1987, 52, 4137–4139 CrossRef; (b) J. B. Hendrickson and M. S. Hussoin, J. Org. Chem., 1989, 54, 1144–1149 CrossRef CAS.
- Z. Nikolovska-Coleska, R. Wang, X. Fang, H. Pan, Y. Tomita, P. Li, P. P. Roller, K. Krajewski, N. G. Saito, J. A. Stuckey and S. Wang, Anal. Biochem., 2004, 332, 261–273 CrossRef CAS PubMed.
- S. Prakash, T. Inobe, A. J. Hatch and A. Matouschek, Nat. Chem. Biol., 2009, 5, 29–36 CrossRef CAS PubMed.
- Y. Li, J. Yang, A. Aguilar, D. McEachern, S. Przybranowski, L. Liu, C. Y. Yang, M. Wang, X. Han and S. Wang, J. Med. Chem., 2019, 62, 448–466 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9cc07795b |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2019 |