
Catalytic selective mono- and difluoroalkylation using fluorinated silyl enol ethers
 *abc
*abc
Abstract
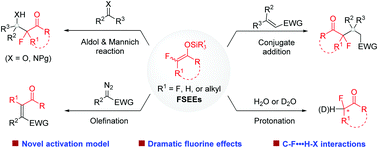
The judicious incorporation of a fluoroalkyl moiety often brings about beneficial effects on the properties of bioactive molecules. Consequently, efficient methods for selective fluoroalkylation are much sought after in drug discovery. Despite significant achievements in trifluoromethylation, selective mono- and difluoroalkylation is still undeveloped. Catalytic functionalization of fluorinated silyl enol ethers (FSEEs) emerges as a fruitful approach for the diversity-oriented synthesis of value-added α-mono or difluoroalkylated ketones. In this feature article, we detail our efforts in developing catalytic selective mono- and difluoroalkylation reactions using FSEEs. Specifically, we highlight our findings such as activating FSEEs by amines for catalytic enantioselective synthesis, taking advantage of the often observed high activity of FSEEs over the non-fluorinated analogues for reaction development, and the influence of C–F⋯H–X interactions on reactivity and selectivity.
 Please wait while we load your content...
Please wait while we load your content...

