
Highly stereoselective intramolecular Buchner reaction of diazoacetamides catalyzed by a Ru(II)–Pheox complex†
Nga
Phan Thi Thanh
 ,
Masaya
Tone
,
Hayato
Inoue
,
Ikuhide
Fujisawa
and
Seiji
Iwasa
*
,
Masaya
Tone
,
Hayato
Inoue
,
Ikuhide
Fujisawa
and
Seiji
Iwasa
*
Department of Applied Chemistry and Life Science, Toyohashi University of Technology, 1-1 Hibarigaoka, Tempaku-cho, Toyohashi, Aichi 441-8580, Japan. E-mail: iwasa@chem.tut.ac.jp
First published on 10th October 2019
Abstract
This work reports the first efficient enantioselective intramolecular Buchner reaction of diazoacetamides. The Ru(II)–Pheox catalyst was shown to be highly efficient in this transformation in terms of both the regio- and enantioselectivity (up to 99% ee) giving the desired products in quantitative yield.
Medium ring-containing organic molecules, such as seven-membered rings, are the cornerstone of many bioactive natural compounds such as guaiane sesquiterpenes, guaianolide sesquiterpene lactones, and diterpene tiglianes.1 However, there are few reports on their synthesis. Unlike five- and six-membered rings, the synthesis of seven-membered rings is more challenging and generally limited to multi-step processes rather than direct intramolecular reactions.2 Thus, development of an efficient method to prepare these scaffolds has attracted a significant amount of research attention.
Over the past few decades, the transition metal-catalyzed intramolecular Buchner reaction3 has been reported by several research groups.4 This unique strategy toward seven-membered carbocycles has been utilized in natural product synthesis.5 However, the catalytic intramolecular reaction of diazoacetamides, diazoketones and diazoesters usually leads to competition between the Buchner and C–H insertion reactions.6 Therefore, many reports deal with controlling the regioselectivity of the reaction, which not only depends on the type of starting material used, but also the nature of the reaction solvent.
Moreover, when compared to the intramolecular C–H insertion reaction of diazoacetamides, there are fewer reports on the Buchner reaction (Scheme 1a and b).7 In particular, very few examples have addressed the stereoselectivity of the Buchner product from the corresponding diazoacetamide.8 To date, only one research study by Doyle and co-workers (2015) has reported the asymmetric intramolecular Buchner reaction of diazoacetamides, whereby N-tert-butyl-N-(p-methoxybenzyl)enoldiazoacetamide resulted in a mixture of the C–H insertion product and Buchner product in a total yield of 90% with moderate enantioselectivities of 41 and 53% ee, respectively (Scheme 1c).8
 | ||
| Scheme 1 Transition metal catalytic carbene transfer reaction of diazoacetamides. | ||
Recently, we have developed a Ru(II)–Pheox9a complex, which is efficient in carbene transfer reactions, in particular, asymmetric cyclopropanation and Si–H insertion reactions.9
Driven by our interests in the catalytic intramolecular Buchner reaction of diazoacetamide, the efficiency displayed by the Ru(II)–Pheox catalyst, and the importance of the seven-membered ring scaffold in natural product synthesis,5 we started to study the enantioselective reaction, which is much more challenging (Scheme 2).
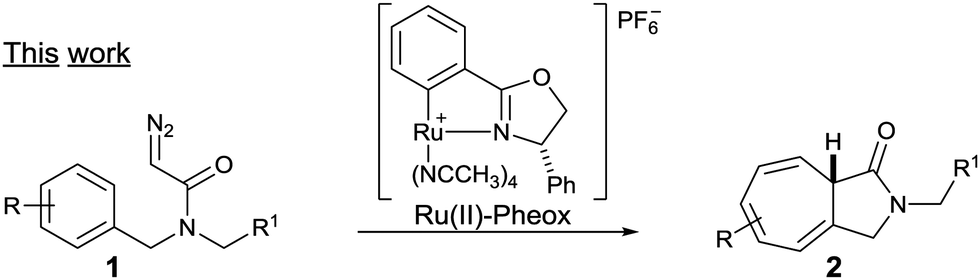 | ||
| Scheme 2 Asymmetric intramolecular reaction of diazoacetamides catalyzed by the Ru(II)–Pheox complex. | ||
Herein, we report the development of an intramolecular Buchner reaction of a variety of N-benzyl diazoamide derivatives in the presence of a chiral Ru(II)–Pheox catalyst. The aromatic rings are converted into the corresponding γ-lactam ring fused seven-membered ring system with high regio- and stereoselectivity.
At the outset of this investigation, N,N-bis(4-methoxybenzyl)-2-diazoacetamide 1b was chosen as the substrate using 1 mol% of catalyst to optimize the reaction conditions. Initially, well-known carbene transfer catalysts were screened and the results summarized in Table 1.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Cat. | Time [min] |
2b![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3ba :3ba |
Yieldb [%] | 2b ee [%] |
|
a The ratio of 2b:3b was determined using 1H NMR spectroscopy.
b Isolated yield.
c Determined using chiral HPLC analysis.
d The reaction temperature was 40 °C.
|
|||||
| 1 | Cat. 1 | 48 h | 100:0 |
52 | 0 |
| 2d | Cat. 2 | 60 | 100:0 |
87 | 15 |
| 3 | Cat. 3 | 2 | 100:0 |
99 | 99 |
| 4 | Cat. 4 | 2 | 100:0 |
95 | 21 |
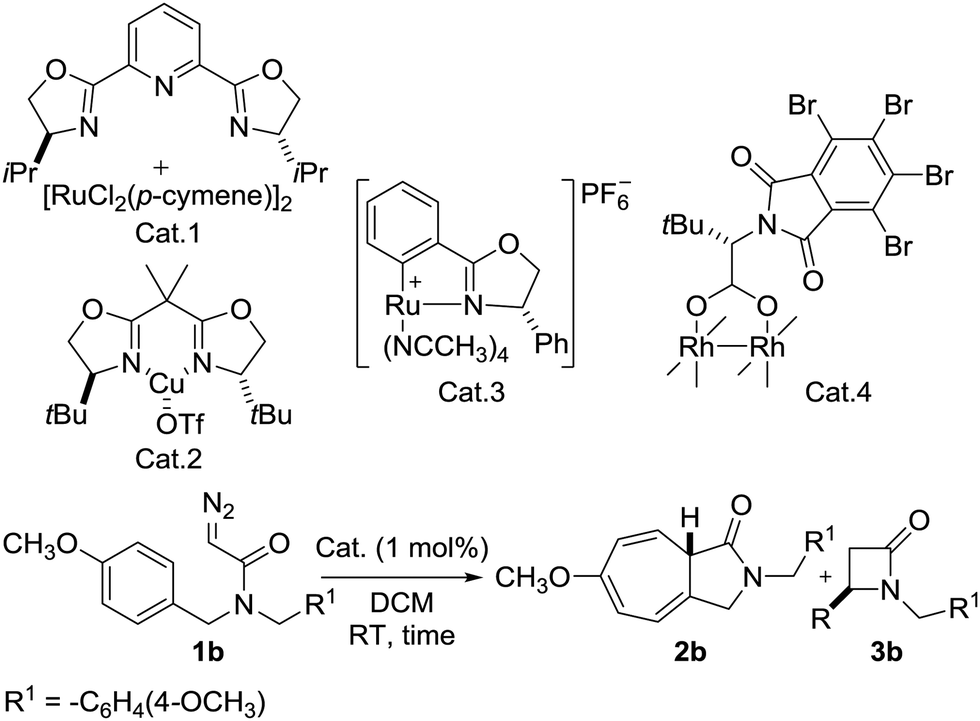
Extensive studies on the reaction conditions indicated that after 48 h, product 2b was obtained in 52% yield with no chirality using Ru(II)–Pybox10 (Table 1, entry 1). When Cu(II)–Box11 was used, product 2b was formed in 87% yield and 15% ee (Table 1, entry 2). In the case of the Rh2(S-TBPTTL)412 complex, the yield of 2b increased dramatically to 95%. However, the enantioselectivity was relatively low (21% ee) (Table 1, entry 4). Screening the various Ru(II)–Pheox catalyst derivatives developed by our group showed that the chiral Ru(II)–Pheox complex (Cat. 3) was the most effective catalyst (Table 1, entry 3 and Table S1, ESI†).9 The reaction proceeds rapidly to give 2b in excellent yield (99%) with almost perfect enantioselectivity (99% ee). For more details on the catalyst screening process, see the ESI.†
We next focused on the efficiency of the Ru(II)–Pheox catalyst and the results shown in Table 2. We found that decreasing the catalyst loading from 1 to 0.002 mol% showed no change in the enantioselectivity (99% ee) of product 2b, while the TON and TOF values increased (Table 2). Using a very small amount of the Ru(II)–Pheox catalyst (0.005 mol%) gave product 2b within 2 min in 99% yield with excellent TOF (9900 min−1) (Table 2, entry 4). When 0.003 mol% of catalyst was used, the TON increased dramatically to 33000 (Table 2, entry 5).
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | X [%] | Time [min] | TONa | TOFb [min−1] | Yieldc [%] | 2b eed [%] |
| a TON = moles of desired product (2b)/moles of catalyst (Ru(II)–Pheox). b TOF = TON/reaction time (min). c Isolated yield. d Determined using chiral HPLC analysis. | ||||||
| 1 | 1 | 2 | 99 | 49.5 | 99 | 99 |
| 2 | 0.1 | 2 | 990 | 495 | 99 | 99 |
| 3 | 0.01 | 2 | 9900 | 4950 | 99 | 99 |
| 4 | 0.005 | 2 | 19800 |
9900 | 99 | 99 |
| 5 | 0.003 | 30 | 33000 |
1100 | 99 | 99 |
| 6 | 0.002 | 60 | 10000 |
167 | 20 | 99 |
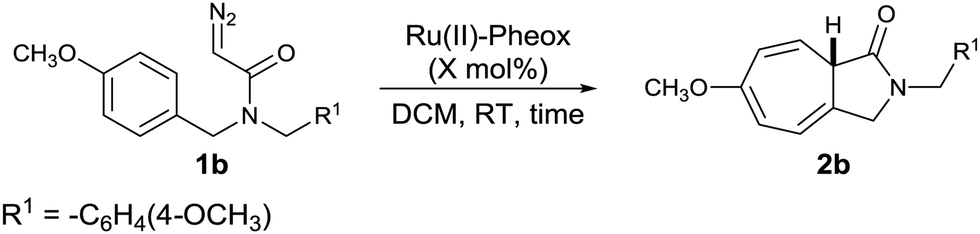
In Table 2, we found that decreasing catalyst loading from 1 to 0.005 mol% Ru(II)–Pheox, the reaction kept proceeding rapidly with excellent regio- and stereoselectivities. Therefore the Ru(II)–Pheox catalyst is extremely efficient for synthesis product 2b from the corresponding diazoacetamide 1b. However, to avoid substrate dependency for various diazoamides, we decided to use 1 mol% Ru(II)–Pheox as the standard condition for screening solvent and substrate scope of the reaction.
In addition, the influence of various solvents on the decomposition of diazoacetamides was examined and the results shown in Table S2 (ESI†). DCM was found to be the best solvent for the Ru(II)–Pheox catalyzed reaction. For more details on the optimization of the reaction conditions, see the ESI.†
Using the optimized reaction conditions, we decided to explore the substrate scope of the reaction (Table 3). Various diazoacetamides of N,N-bis(aryl)-2-diazo-acetamides were examined (Table 3, entries 1–7). Substrates bearing either electron-withdrawing or electron-donating groups (R = H, F, Cl, Br, CH3, and OCH3) on the N-benzyl ring were tolerated in the reaction, giving the desired products (2a–g) in 69–99% yield and 74–99% ee.
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | 1 | R | R1 | Time [min] |
2:3a |
2 Yieldb [%] | 2 eec [%] |
| a The ratio was determined using 1H NMR spectroscopy of the reaction mixture. b Isolated yield. c Determined using chiral HPLC analysis. | |||||||
| 1 | 1a | H | C6H5 | 2 | 75:25 |
69 | 78 |
| 2 | 1b | 4-OCH3 | 4-CH3OC6H4 | 2 | 100:0 |
99 | 99 |
| 3 | 1c | 4-CH3 | 4-CH3C6H4 | 2 | 97:3 |
96 | 97 |
| 4 | 1d | 4-Cl | 4-ClC6H4 | 2 | 83:17 |
80 | 96 |
| 5 | 1e | 4-Br | 4-BrC6H4 | 2 | 80:20 |
70 | 95 |
| 6 | 1f | 4-F | 4-FC6H4 | 2 | 92:8 |
91 | 90 |
| 7 | 1g | 3-OCH3 | 3-CH3OC6H4 | 2 | 100:0 |
87 | 74 |
| 8 | 1i | H | H | 4 h | 45:55 |
40 | 71 |
| 9 | 1j | 4-OCH3 | H | 2 | 100:0 |
76 | 99 |
| 10 | 1k | 4-CH3 | H | 2 | 90:10 |
48 | 99 |
| 11 | 1k | 4-CH3 | H | 4 h | 93:7 |
67 | 99 |
| 12 | 1l | 4-Cl | H | 4 h | 83:17 |
61 | 92 |
| 13 | 1m | 4-Br | H | 4 h | 80:20 |
43 | 96 |
| 14 | 1n | 4-F | H | 4 h | 79:21 |
55 | 92 |
| 15 | 1o | 4-NO2 | H | 4 h | — | n.o. | — |
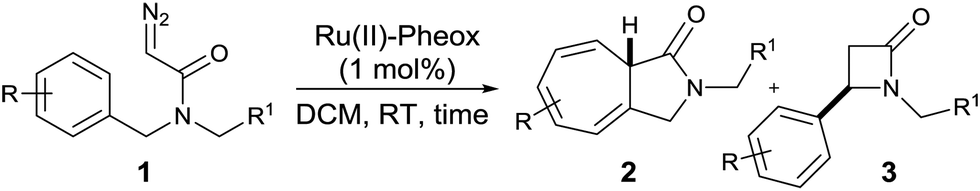
Substitution with an electron-donating group (e.g., 4-OCH3, 3-OCH3, and 4-CH3) on the N-benzyl ring moiety has a strong impact on the reaction (Table 3, entries 2, 3 and 7). The corresponding Buchner reaction products were obtained in excellent yield (up to 99%) and enantioselectivity (up to 99% ee). In the case of substrates bearing an electron-withdrawing group (namely 4-Cl, 4-Br and 4-F), the rate of the Buchner reaction slightly decreased and formation of the C–H insertion product was observed (Table 3, entries 4–6). Nevertheless, the yield and enantioselectivity of the products (2d–f) remained excellent (70–91% yield and 90–96% ee). In addition, the bicyclic product 2d was prepared with the purpose of growing crystals suitable for analysis. The structure of 2d was confirmed and the absolute configuration was determined to be the S configuration using single-crystal X-ray diffraction (Fig. 1 and Fig. S1, ESI†).
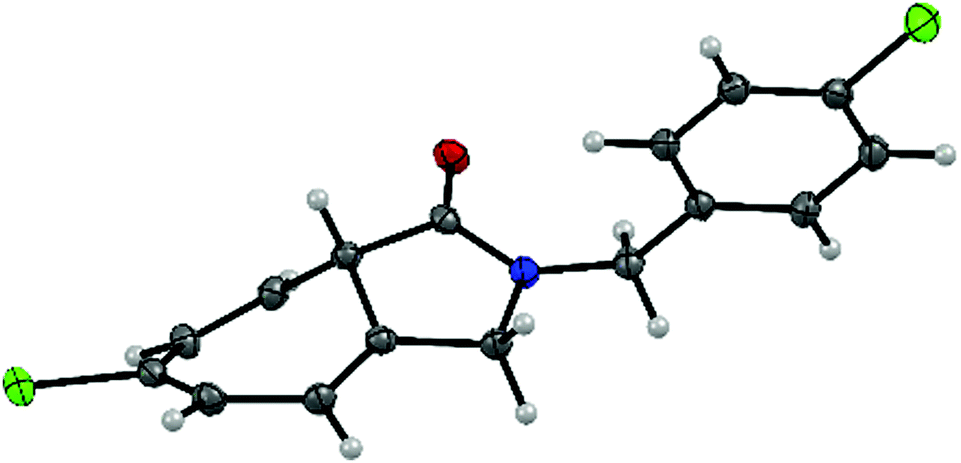 | ||
| Fig. 1 X-ray analysis of (S)-6-chloro-2-(4-chlorobenzyl)-3,8a-dihydrocyclohepta[c]pyrrol-1(2H)-one (2d). | ||
In short, entries 1–7 in Table 3 present an overview of the decomposition of a series of N,N-bis(aryl)-2-diazo-acetamides used to prepare the target seven-membered ring products (2a–g) with excellent stereo- and regioselectivity.
Besides, a diazoamide bearing both electron-withdrawing and electron-donating groups (1h) was also investigated as a substrate, affording the desired Buchner reaction product (2h) in high yield (84%) and excellent enantioselectivity (99% ee) (Scheme 3).
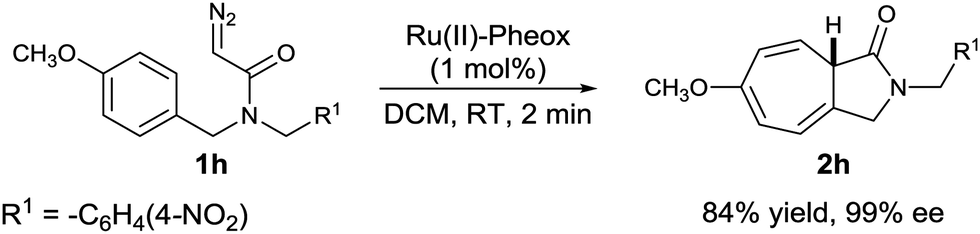 | ||
| Scheme 3 Asymmetric intramolecular reaction of diazoacetamide 1h catalyzed by Ru(II)–Pheox. | ||
Subsequently, a series of N-aryl-2-diazo-N-methylacetamides was also investigated under the same reaction conditions (Table 3, entries 8–15). Substrates with both electron-withdrawing and electron-donating groups work well in the reaction and afford their Buchner reaction products (2i–n) in moderate to good yield (40–76%) and enantioselectivity (71–99% ee).
The reaction afforded the intramolecular Buchner product 2j (R = 4-OCH3) in 76% yield with high enantioselectivity (99% ee) (Table 3, entry 9). Switching the substrate to 1k (R = 4-CH3) dramatically changed the reaction, affording the corresponding seven-membered ring product (2k) in 48% yield in 2 min (Table 3, entry 10). Surprisingly, we found that the reaction could afford product 2k in 67% yield over a longer reaction time (4 h) (Table 3, entry 11). The dimerization reaction was prevented and the reaction yield was improved upon slow addition of a solution of the diazoacetamide to a stirred mixture of the Ru(II)–Pheox catalyst in DCM over 4 h (Table 3, entries 8 and 11–15).
Furthermore, there is intense competition between the reactive sites of the N-aryl-2-diazo-N-methylacetamide (Table 3, entries 8 and 12–14). Therefore, bicyclic products 2i and 2l–2n could be obtained in moderate yield (40–61%). In the case of diazo compound 1o, the corresponding product 2o was not obtained.
As a plausible explanation, the substituent changes the electronic properties of the benzene ring and affects the regioselectivity. Nucleophilic substituents, such as 4-CH3 and 4-OCH3, are regarded as electronic donating groups, which increase the electropositivity of the aryl group and improve the reactivity in the aromatic addition reaction. Electrophilic substituents, such as –Cl, –Br, –F, and –H, are regarded as electron-withdrawing groups, which decrease the electropositivity of the aryl group and favor the C–H insertion reaction.
In summary, we have presented a highly stereoselective intramolecular Buchner reaction of diazoacetamides using a Ru(II)–Pheox catalyst. Specifically, a variety of γ-lactam fused 5,7-bicyclic-heptatriene derivatives have been prepared from diazoacetamides in up to 99% yield with high enantioselectively (up to 99% ee) using a chiral Ru(II)–Pheox catalyst under mild reaction conditions. The product containing diene can be used for further transformation via the Diels–Alder cycloaddition reaction.7a,13
This work was supported by a Grant-in-Aid for Scientific Research (B) (no. 26288087) and Ikeda Bussan Co. Ltd.
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) E. Rodriguez, G. H. N. Towers and J. C. Mitchell, Phytochemistry, 1976, 15, 1573–1580 CrossRef CAS; (b) W. He, M. Cik, G. Appendino, L. Puyvelde, J. E. Leysen and N. Kimpe, Mini-Rev. Med. Chem., 2002, 2, 185–200 CrossRef CAS PubMed; (c) H. B. Wang, X. Y. Wang, L. P. Liu, G. W. Qin and T. G. Kang, Chem. Rev., 2015, 115, 2975–3011 CrossRef CAS PubMed.
- (a) M. A. Battiste, P. M. Pelphrey and D. L. Wright, Chem. – Eur. J., 2006, 12, 3438–3447 CrossRef CAS PubMed; (b) H. Butenschon, Angew. Chem., Int. Ed., 2008, 47, 5287–5290 CrossRef PubMed; (c) X. Li, R. E. Kyne and T. V. Ovaska, J. Org. Chem., 2007, 72, 6624–6627 CrossRef CAS PubMed.
- E. Buchner and T. Curtius, Ber. Dtsch. Chem., 1885, 18, 2377–2379 CrossRef.
- (a) H. Duddeck, M. Kennedy, M. A. Mckervey and F. M. Twohig, J. Chem. Soc., Chem. Commun., 1988, 24, 1586–1588 RSC; (b) M. P. Doyle, D. G. Ene, D. C. Forbes and T. H. Pillow, Chem. Commun., 1999, 1691–1692 RSC; (c) A. Padwa, D. J. Austin, A. T. Price, M. A. Semones, M. P. Doyle, M. N. Protopopova, W. R. Winchester and A. Tran, J. Am. Chem. Soc., 1993, 115, 8669–8680 CrossRef CAS; (d) A. A. Cordi, J. M. Lacoste and P. Hennig, J. Chem. Soc., Perkin Trans. 1, 1993, 3–4 RSC; (e) C. A. Merlic, A. L. Zechman and M. M. Miller, J. Am. Chem. Soc., 2001, 123, 11101–11102 CrossRef CAS PubMed; (f) M. P. Doyle, W. Hu and D. J. Timmons, Org. Lett., 2001, 3, 933–935 CrossRef CAS PubMed; (g) M. P. Doyle and I. M. Phillips, Tetrahedron Lett., 2001, 42, 3155–3158 CrossRef CAS; (h) A. R. Maguire, P. O’Leary, F. Harrington, S. E. Lawrence and A. J. Blake, J. Org. Chem., 2001, 66, 7166–7177 CrossRef CAS PubMed; (i) J. L. Kane, K. M. Shea, A. L. Crombie and R. L. Danheiser, Org. Lett., 2011, 13, 1081–1084 Search PubMed; (j) O. A. McNamara, N. R. Buckley, P. O’Leary, F. Harrington, N. Kelly, S. O’Keeffe, A. Stack, S. O’Neill, S. E. Lawrence, C. N. Slattery and A. R. Maguire, Tetrahedron, 2014, 70, 6870–6878 CrossRef CAS; (k) G. S. Fleming and A. B. Beeler, Org. Lett., 2017, 19, 5268–5271 CrossRef CAS PubMed; (l) S. O’Keeffe, F. Harrington and A. R. Maguire, Synlett, 2007, 2367–2370 Search PubMed; (m) S. O’Neill, S. O’Keeffe, F. Harrington and A. R. Maguire, Synlett, 2009, 2312–2314 Search PubMed; (n) C. N. Slattery, L.-A. Clarke, S. O’Neill, A. Ring, A. Forda and A. R. Maguire, Synlett, 2012, 765–767 CAS; (o) D. C. Crowley, D. Lynch and A. R. Maguire, J. Org. Chem., 2018, 83, 3794–3805 CrossRef CAS PubMed.
- B. Frey, A. P. Wells, D. H. Rogers and L. N. Mander, J. Am. Chem. Soc., 1998, 120, 1914–1915 CrossRef CAS.
- For the selected intramolecular reaction of diazoacetamides, see: (a) P. M. Doyle, M. S. Shanklin, S. M. Oon, H. Q. Pho, F. R. van der Heide and W. R. Veal, J. Am. Chem. Soc., 1988, 53, 3384–3386 Search PubMed; (b) M. P. Doyle, R. J. Pieters, J. Taunton and H. Q. Pho, J. Org. Chem., 1991, 56, 820–829 CrossRef CAS; (c) H. C. Yoon, J. M. Zaworoko, B. Moulton and W. K. Jung, Org. Lett., 2001, 22, 3539–3542 CrossRef PubMed; (d) W. H. Cheung, S. L. Zheng, G. C. Zhou and C. M. Che, Org. Lett., 2003, 5, 2535–2538 CrossRef CAS PubMed; (e) Z. Chen, Z. Chen, Y. Jiang and W. Hu, Synlett, 2004, 1763–1764 CAS; (f) K. W. Choi, W. Y. Yu and C. M. Che, Org. Lett., 2005, 7, 1081–1084 CrossRef PubMed; (g) L. D. Flanigan, H. C. Yoon and W. K. Jung, Tetrahedron Lett., 2005, 46, 143–146 CrossRef; (h) M. Grohman, S. Buck, L. Schaffler and G. Mass, Adv. Synth. Catal., 2006, 348, 2203–2211 CrossRef; (i) M. Grohmann, S. Buck, L. Schäffler and G. Mass, Tetrahedron, 2007, 63, 12172–12178 CrossRef CAS; (j) F. R. L. Gomes, F. A. Trindade, R. N. Candeias, M. P. Gois and A. M. C. Afonso, Tetrahedron Lett., 2008, 49, 7372–7375 CrossRef; (k) M. E. Zakrzewska, P. M. S. D. Cal, N. R. Candeias, R. B. Lukasik, C. A. M. Afonso, M. N. Ponte and P. M. P. Gois, Green Chem. Lett. Rev., 2012, 5, 211–240 CrossRef CAS; (l) X. Xu, Y. Deng, N. D. Yim, Y. P. Zavalij and P. M. Doyle, Chem. Sci., 2015, 6, 2196–2201 RSC; (m) Y. Deng, C. Jing, H. Arman and M. P. Doyle, Organometallics, 2016, 35, 3413–3420 CrossRef CAS; (n) H. Li, X. Ma and M. Lei, Dalton Trans., 2016, 1–3 Search PubMed ; For the selected intramolecular reaction of diazoesters, see: ; (o) P. M. Doyle, L. J. Westrum, W. N. E. Wolthuis, M. M. See, W. P. Boone, V. Bagheri and M. M. Pearson, J. Am. Chem. Soc., 1993, 115, 958–964 CrossRef; (p) P. M. Doyle, A. V. Kalinin and D. G. Ene, J. Am. Chem. Soc., 1996, 118, 8837–8846 CrossRef ; For the selected intramolecular reaction of diazoketones, see: ; (q) A. Padwa, G. E. Fryxell and L. Zhi, J. Am. Chem. Soc., 1990, 112, 3100–3109 CrossRef CAS; (r) J. P. Snyder, A. Padwa, T. Stengel, A. J. Arduengo, A. Jockisch and H. J. Kim, J. Am. Chem. Soc., 2001, 123, 11318–11319 CrossRef CAS PubMed; (s) S. E. Reisman, R. R. Nani and S. Levin, Synlett, 2011, 2437–2442 CrossRef CAS; (t) P. A. McDowell, D. A. Foley, P. Oleary, A. Ford and A. R. Maguire, J. Org. Chem., 2012, 77, 2035–2040 CrossRef CAS PubMed; (u) T. Ye and M. A. McKervey, Chem. Rev., 1994, 94, 1091–1160 CrossRef CAS; (v) A. Ford, H. Miel, A. Ring, C. N. Slattery, A. R. Maguire and M. A. McKervey, Chem. Rev., 2015, 115, 9981–10080 CrossRef CAS PubMed.
- (a) C. J. Moody, S. Miah, A. M. Z. Slawin, D. J. Mansfield and I. A. Richards, J. Chem. Soc., Perkin Trans. 1, 1998, 4067–4075 RSC; (b) S. Mo and J. Xu, ChemCatChem, 2014, 6, 1679–1683 CrossRef CAS; (c) S. Mo, X. Li and J. Xu, J. Org. Chem., 2014, 79, 9186–9195 CrossRef CAS PubMed; (d) J. Liu, J. Tu, Z. Yang, C. Pak and J. Xu, Tetrahedron, 2017, 73, 4616–4626 CrossRef CAS.
- X. Xu, Y. Dang, D. N. Yim, P. Y. Zavalij and M. P. Doyle, Chem. Sci., 2015, 6, 2196–2201 RSC.
- For synthesis and characterization of catalysts Ru(II)–Pheox, see: (a) A. M. Abu-Elfotoh, K. Phomkeona, K. Shibatomi and S. Iwasa, Angew. Chem., Int. Ed., 2010, 49, 8439–8443 CrossRef CAS PubMed ; For selected Ru(II)–Pheox catalyzed reaction, see: ; (b) S. Chanthamath and S. Iwasa, Acc. Chem. Res., 2016, 49, 2080–2090 CrossRef CAS PubMed; (c) Y. Nakagawa, S. Chanthamath, I. Fujisawa, K. Shibatomi and S. Iwasa, Chem. Commun., 2017, 53, 3753–3756 RSC; (d) Y. Nakagawa, Y. Imokawa, I. Fujisawa, N. Nakayama, H. Goto, S. Chanthamath, K. Shibatomi and S. Iwasa, ACS Omega, 2018, 3, 11286–11289 CrossRef CAS PubMed; (e) M. Tone, Y. Nakagawa, S. Chanthamath, I. Fujisawa, N. Nakayama, H. Goto, K. Shibatomi and S. Iwasa, RSC Adv., 2018, 8, 39865–39869 RSC; (f) Y. Nakagawa, N. Nakayama, H. Goto, I. Fujisawa, S. Chanthamath, K. Shibatomi and S. Iwasa, Chirality, 2019, 31, 52–61 CrossRef CAS PubMed; (g) Y. Nakagawa, S. Chanthamath, Y. Liang, K. Shibatomi and S. Iwasa, J. Org. Chem., 2019, 84, 2607–2618 CrossRef CAS PubMed; (h) C. T. L. Le, S. Ozaki, S. Chanthamath, K. Shibatomi and S. Iwasa, Org. Lett., 2018, 20, 4490–4494 CrossRef CAS PubMed; (i) L. D. Ho, N. Otog, I. Fujisawa and S. Iwasa, Org. Lett., 2019, 21, 7470–7474 CrossRef PubMed.
- H. Nishiyama, Y. Itoh, Y. Sugawara, H. Matsumoto, K. Aoki and K. Itoh, Bull. Chem. Soc. Jpn., 1995, 68, 1247–1262 CrossRef CAS.
- D. A. Evans, K. A. Woerpel, M. M. Hinman and M. M. Faul, J. Am. Chem. Soc., 1991, 113, 726–728 CrossRef CAS.
- (a) K. Goto, K. Takeda, M. Anada, K. Ando and S. Hashimoto, Tetrahedron Lett., 2011, 52, 4200–4203 CrossRef; (b) T. Goto, K. Takeda, N. Shimada, H. Nambu, M. Anada, M. Shiro, K. Ando and S. Hashimoto, Angew. Chem., Int. Ed., 2011, 50, 6803–6808 CrossRef CAS PubMed; (c) F. G. Adly and A. Ghanem, Chirality, 2014, 26, 692–711 CrossRef CAS PubMed.
- (a) A. R. Maguire, N. R. Buckley, P. O’Leary and G. Ferguson, Chem. Commun., 1996, 2595–2596 RSC; (b) S. S. Templin, N. J. Wallock, D. W. Bennett, T. Siddiquee, D. T. Haworth and W. A. Donaldson, J. Heterocycl. Chem., 2007, 44, 719–723 CrossRef CAS; (c) A. Saba, Tetrahedron Lett., 1990, 31, 4657–4660 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1944984. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9cc06889a |
| This journal is © The Royal Society of Chemistry 2019 |