
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIridium(III) complexes of 1,2,4-triazines as potential bioorthogonal reagents: metal coordination facilitates luminogenic reaction with strained cyclooctynes†
Valery N.
Kozhevnikov
 *a,
Michael E.
Deary
b,
Theodora
Mantso
a,
Mihalis I.
Panayiotidis‡
a and
Mark T.
Sims
a
*a,
Michael E.
Deary
b,
Theodora
Mantso
a,
Mihalis I.
Panayiotidis‡
a and
Mark T.
Sims
a
aDepartment of Applied Sciences, Northumbria University, Newcastle upon Tyne, NE1 8ST, UK. E-mail: valery.kozhevnikov@northumbria.ac.uk
bDepartment of Geography and Environmental Sciences, Northumbria University, Newcastle upon Tyne, NE1 8ST, UK
First published on 1st November 2019
Abstract
In this paper we describe unprecedented Ir(III) complexes of 5-(2-pyridyl)-1,2,4-triazine and their reactivity towards the strained cyclooctyne BCN. The coordination of a 1,2,4-triazine ring to an iridium(III) ion drastically increases the speed of the reaction, showing the second order rate constant of 8 M−1 s−1, the record value to date for a triazine-BCN reaction.
Bioorthogonal reactions occur in living systems without interfering with intracellular processes and have found widespread applications in biological sciences.1 The success of bioorthogonal chemistry is underpinned by the development of click reactions and reagents that are designed to selectively react with each other but not to react with any of the naturally present molecules. Although this requirement is quite challenging, the toolkit of bioorthogonal chemistry includes many reactions. One of the fastest bioorthogonal reactions is the Inverse Electron Demand Diels Alder (IEDDA) reaction of 1,2,4,5-tetrazines with strained alkenes and alkynes.2 These reactions have now found widespread applications in bioimaging and in a variety of other fields.3 However, the most reactive tetrazines are also the least stable under physiological conditions. Recently, 1,2,4-triazines have been reported as potential bioorthogonal reagents offering excellent stability.4–11 Unfortunately, 1,2,4-triazines react much slower in IEDDA reactions. Although it has been shown that the kinetics can be improved by the introduction of electron-withdrawing groups,9,10 the rate constant is two-three orders of magnitude lower than for tetrazines.
Metal ions often catalyse IEDDA reactions by influencing the frontier orbitals of the reactants and minimising the activation energy of the reaction. For example Lo and co-workers have recently shown that Ir(III) complexes of tetrazines react much faster in IEDDA reactions than uncoordinated ligands.12 This phenomenon inspired us to design metal complexes of 1,2,4-triazines to improve the kinetics of the IEDDA reactions. In addition, the result of the IEDDA reaction would be a polypyridine type metal complex that often has attractive properties. For example, we chose cyclometallated iridium(III) complexes because they are among the brightest emitters used in bioimaging and bioorthogonal chemistry.13,14,25 These complexes are easy to prepare and photo-stable, and their photophysical properties, such as absorption and emission, are highly tunable. The long-lived emission of these complexes provides an opportunity for the time-delayed elimination of background luminescence and for phosphorescent lifetime imaging.15–23 In this paper we report unprecedented Ir(III) complexes of 1,2,4-triazines as luminogenic bioorthogonal reagents that react with BCN much faster than the uncoordinated 1,2,4-triazine ligand approaching the kinetic values of widely used tetrazines. However, the relatively high toxicity of the complexes imposes some limitations to bioorthogonality and further studies are necessary to address this problem.
The key feature of our complexes is that the 1,2,4-triazine ring is directly coordinated to the metal centre. Because during the IEDDA reaction, the N1 and N2 atoms of the 1,2,4-triazine ring are lost, the coordination to the metal should be through the N4 atom. Only two isomers satisfy this criterion, and among these, 5-(pyridine-2-yl)-1,2,4-triazine 1 is the best choice because competitive coordination to the N2 atom of the triazine ring is avoided (Fig. 1).
 | ||
| Fig. 1 Possible (pyridine-2-yl)-1,2,4-triazines with green and red arrows showing desirable and undesirable coordination respectively. Only 5-(pyridine-2-yl)-1,2,4-triazine 1 avoids undesirable coordination and was chosen as the most suitable ligand. | ||
It should be noted that the ligand 1 has not been previously reported and generally examples of N5 coordination are very rare in the literature. Additional motivation for the use of isomer 1 is that 5-substituted 1,2,4-triazines are the least sterically crowded and able to react even with sterically encumbered strained alkynes such as tetramethylthiacycloheptyne (TMTH).5 To prepare the ligand 1, we first tested the most direct route by reacting 2-pyridineglyoxal 4 with formamidrazone24 but unfortunately, the reaction gave a mixture of two isomers 1 and 3 that we could not separate either by recrystallization, or by column chromatography. We therefore used another synthetic route, depicted in Scheme 1. The route started with the reaction of pyridine glyoxal 4 with thiosemicarbazide 5 to give selectively 3-mercapto triazine 6, which was then converted into the hydrazine derivative 7, followed by dehydrazination by treatment with sodium methoxide to give the desired ligand 1. Although the overall yield was only 18%, this route is preferable because only the desired 5-(2-pyridyl)-1,2,4-triazine 1 forms during the reaction. The iridium(III) complex 8Cl was then prepared by the treatment of 1 with dichloro-bridged intermediate [Ir(dfppy)2Cl]2. It should be noted that many other dichloro-bridged Ir(III) precursors can be used at this stage, enabling the tuning of photophysical properties, solubility and biocompatibility of the complexes.13,14 The chloride salt 8Cl can be easily converted into a more hydrophobic hexafluorophosphate analogue 8PF6 by treatment with excess sodium hexafluorophosphate. While hexafluorophosphate salts are easier to purify, chloride derivatives provide much better solubility in water, a desirable feature for biological applications.
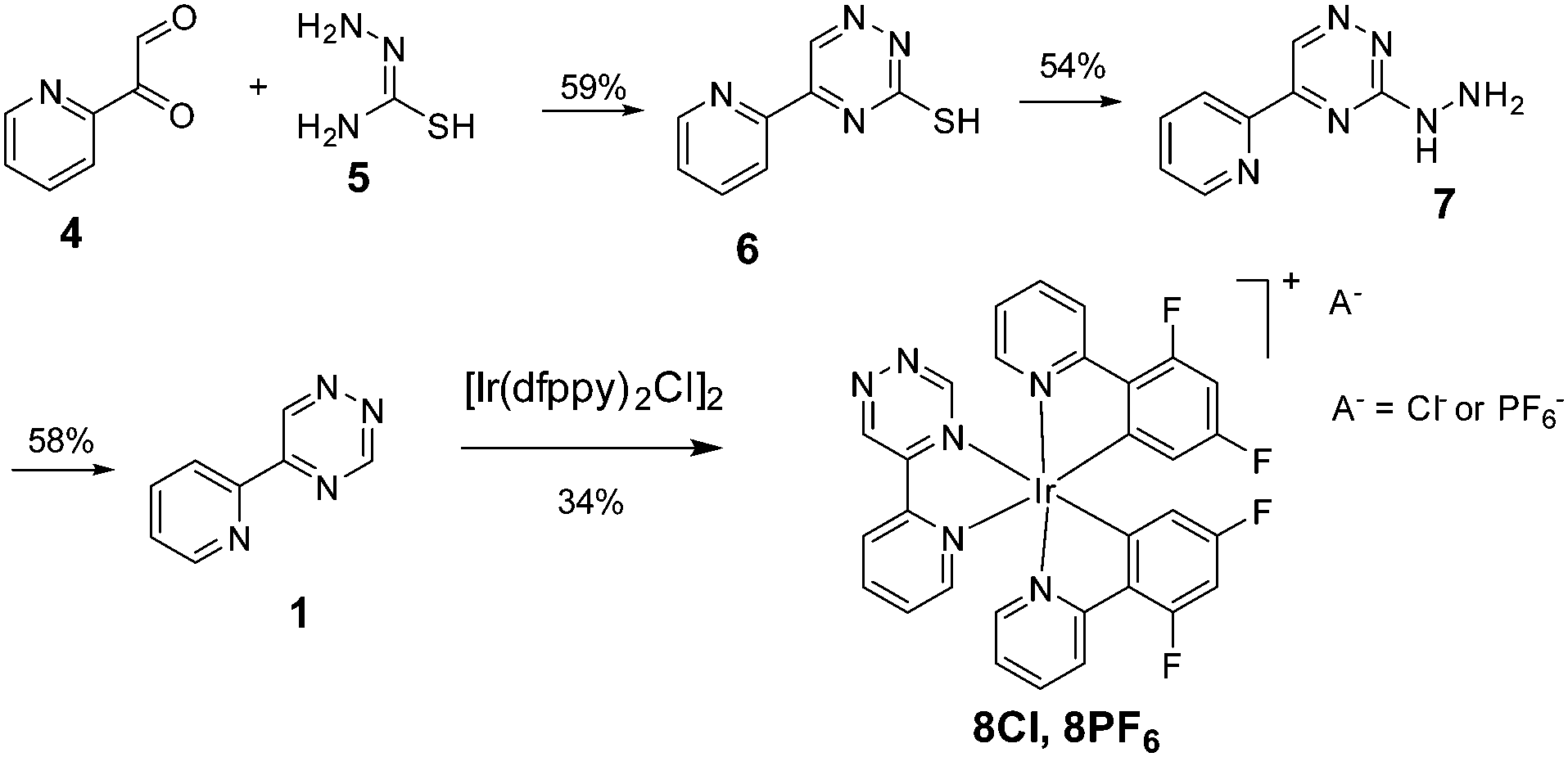 | ||
| Scheme 1 The synthesis of 5-(pyridine-2-yl)-1,2,4-triazine 1 and its iridium(III) complexes 8Cl and 8PF6. | ||
In the cellular environment, the concentrations of biomolecules are very low, demanding that the bioorthogonal reactions should be very fast. For in vivo applications, the rate constant should be in the order of 1 M−1 s−1 or higher.26 The reactions of 1,2,4-triazines with BCN show much slower reaction rates.6 For example, we found that ligand 1 reacts with BCN in methanol at 25 °C with a second order rate constant of k2 = 0.059 M−1 s−1. However, to our delight, we also found that the coordination of the Ir(III) ion to the N4 atom of 1,2,4-triazine increases the second order rate constant by more than two orders of magnitude and the complex 8 shows the rate constant of approximately 8 M−1 s−1 (Table 1). This is a very important discovery that places the metal complexes of 1,2,4-triazines kinetically in the same league as commonly used tetrazines.
| Compound | k 2/M−1 s−1 |
|---|---|
| 1 | 0.059 ± 0.001 |
| 8Cl | 7.870 ± 0.325 |
| 8PF6 | 8.223 ± 0.307 |
In order to understand the influence of metal coordination on the rate of the reaction, we performed computational analysis. Calculated transition-state geometries for the reactions of ligand 1 and complex 8 with BCN were found to exhibit a similar distortion of the diene and similar positioning of the alkyne C atoms of BCN ca. 2 Å from the C3 and C6 atoms of the triazine ring. An example structure showing these features is shown in Fig. 2 for complex 8 (Scheme 2).
 | ||
| Fig. 2 The lowest-energy transition state geometry calculated for the reaction of 8 with BCN, calculated at the M06/6-311G(d,p)/LANL2DZ level. | ||
 | ||
| Scheme 2 Reactions of iridium(III) complexes 8Cl or 8PF6 with BCN and BCN-C10. | ||
Calculated Gibbs activation energies for reactions of 1 and 8 with BCN are given in Table 2. Also listed are the experimental energy barriers determined from the observed rate constants. The quantitative match between the calculated and experimental values is similar to that reported in previous work studying organic species, and a similar trend is exhibited of overestimating the energy barriers when the activation energies are higher.27–29 Significantly, the calculations show a marked decrease in activation energy on going from the free triazine ligand to the Ir coordinated triazine, consistent with the experimental values.
| Diene | ΔG‡calc/kcal mol−1 | ΔG‡exp/kcal mol−1 |
|---|---|---|
| 1 | 26.4 | 19.1 |
| 8 | 20.7 | 16.2 |
Previous computational investigations of organic bioorthogonal reactions have shown the distortion–interaction model to be a valuable tool in the interpretation of experimental rate constants.4,27–30 In this model, individual energies of the distortion of the reactant molecules from their minimum-energy geometries to their transition state geometries are determined, as well as the difference between the energies of the separated reactant transition state geometries and that of the transition state itself. Results of these analyses carried out for reactions of BCN with 1 and 8 are shown in Fig. 2.
The calculated distortion energies of the triazines are both significantly higher than those of BCN for the two transition states, and there is only a small difference between the distortion energies in the two reactions. In contrast, there is a large difference between the calculated interaction energies for the reactions, attributable to the lower calculated frontier orbital energy of complex 8 (−5.109 eV) than the ligand 1 (−1.533 eV), making it much closer to the energy of the BCN HOMO (−7.197 eV) (Fig. 3).
 | ||
| Fig. 3 Calculated distortion energies, Edist, and interaction energies, Eint, for reactions of ligand 1 and complex 8 with BCN, averaged over each of the transition state geometries for each reaction. | ||
Complex 8 shows a luminogenic response to reaction with BCN. In bioimaging, the luminogenic probes that “switch on” only after the conjugating click reaction takes place are particularly important because the luminescent background of the non-conjugated probe is not present and thus there is no need for laborious and time-consuming washing steps. 8PF6 showed a strong luminogenic behaviour in reaction with BCN-C10. While the 8PF6 is virtually non-emissive in acetonitrile solution, the addition of BCN-C10 leads to the appearance of intense luminescence (Fig. 4B). The absorption also changes, which can be registered visually as a transition from red to yellow colour of the solution. The product of the IEDDA reaction is the iridium bipyridine complex 9. Such complexes are well-studied systems20 and by assuming that aliphatic substitution does not drastically change the photophysical properties, one can predict that the quantum yield of the “switched-on” product 9 will be in the region of 10–25% depending on the solvent used. In general, the absorption and emission of cyclometallated Ir(III) complexes such as 9 is highly tunable and can be adjusted to desired values by the use of different sets of ligands. In-depth photophysical investigation and tuning of these systems will be reported in due course.
 | ||
| Fig. 4 Visual appearance of acetonitrile solution of 8PF6 in (A) daylight and (B) under UV (360 nm) irradiation before (left tube) and after (right tube) the reaction with BCN-C10. | ||
Because complexes can be potentially used in in vivo applications, it is important to assess their cytotoxicity. The cytotoxicity profile of 8Cl was studied by performing the Alamar blue cell viability assay in human malignant melanoma (A375) and keratinocyte (HaCaT) cell lines. 8Cl was shown to be cytotoxic in a dose-dependent manner at concentrations ranging from 5 to 100 μM over various incubation periods (24, 48, 72 h) in both cell lines. Overall, HaCaT cells appeared to be more sensitive to 8Cl when compared to A375 cells. The corresponding EC50 values (half maximal effective concentration) for different incubation periods were calculated and shown to be 32.9, 22.8 and 20.7 μM for A375 cells, while they were considerably lower for HaCaT cells at 17.5, 10.2 and 7.3 μM for 24, 48 and 72 h respectively. High toxicity does not automatically mean that the complexes are not suitable for in vivo biorthogonal experiments, rather that care has to be taken to carry out such experiments using concentrations that are nontoxic for the cells. Cytotoxicity can also be significantly reduced in Ir complexes by ligand modifications. For example it was shown that the introduction of oligoethylene glycol chains significantly reduces cytotoxicity.14 While somewhat a barrier to bioorthogonality, high cytotoxicity can potentially be exploited in the development of clickable anticancer drugs. We are currently using other tumor cell lines to demonstrate the cytotoxic effect of the complexes before and after click reaction in experimental models of skin cancer and other solid tumors.
In summary, in this communication we have presented the first example of 1,2,4-triazine metal complexes as dienophiles in IEDDA reactions. We showed that the coordination to the Ir(III) ion drastically increases the rate of the reaction with BCN. Computational analysis indicates that complexation lowers the energy of the reacting orbital, resulting in a more favourable interaction between the reactants that lowers the activation energy. Because the ligands as well as the metal part of the molecule can be varied, a great variety of 1,2,4-triazine metal complexes can be prepared and their properties can be carefully tuned for a particular application. This novel class of click reagents is thus of interest to the field of bioorthogonal chemistry and other scientific fields where metal-based functionalisation is required.
Conflicts of interest
There are no conflicts to declare.References
- M. Boyce and C. R. Bertozzi, Nat. Methods, 2011, 8, 638–642 CrossRef CAS PubMed.
- E. Kozma, O. Demeter and P. Kele, ChemBioChem, 2017, 18, 486–501 CrossRef CAS PubMed.
- B. L. Oliveira, Z. Guo and G. J. L. Bernardes, Chem. Soc. Rev., 2017, 46, 4895–4950 RSC.
- D. N. Kamber, Y. Liang, R. J. Blizzard, F. Liu, R. A. Mehl, K. N. Houk and J. A. Prescher, J. Am. Chem. Soc., 2015, 137, 8388–8391 CrossRef CAS PubMed.
- D. N. Kamber, S. S. Nguyen, F. Liu, J. S. Briggs, H.-W. Shih, R. D. Row, Z. G. Long, K. N. Houk, Y. Liang and J. A. Prescher, Chem. Sci., 2019, 10, 9109–9114 RSC.
- K. A. Horner, N. M. Valette and M. E. Webb, Chem. – Eur. J., 2015, 21, 14376–14381 CrossRef CAS PubMed.
- K. Peewasan and H.-A. Wagenknecht, ChemBioChem, 2017, 18, 1473–1476 CrossRef CAS PubMed.
- U. Reisacher, D. Ploschik, F. Rönicke, G. B. Cserép, P. Kele and H.-A. Wagenknecht, Chem. Sci., 2019, 10, 4032–4037 RSC.
- S. J. Siegl, R. Dzijak, A. Vazquez, R. Pohl and M. Vrabel, Chem. Sci., 2017, 8, 3593–3598 RSC.
- Z. Chen, N. Ren, X. Ma, J. Nie, F.-G. Zhang and J.-A. Ma, ACS Catal., 2019, 9, 4600–4608 CrossRef CAS.
- M. Baalmann, M. J. Ziegler, P. Werther, J. Wilhelm and R. Wombacher, Bioconjugate Chem., 2019, 30, 1405–1414 CrossRef CAS PubMed.
- T. S.-M. Tang, H.-W. Liu and K. K.-W. Lo, Chem. Commun., 2017, 53, 3299–3302 RSC.
- C. Caporale and M. Massi, Coord. Chem. Rev., 2018, 363, 71–91 CrossRef CAS.
- A. M.-H. Yip and K. K.-W. Lo, Coord. Chem. Rev., 2018, 361, 138–163 CrossRef CAS.
- K. K.-W. Lo, B. T.-N. Chan, H.-W. Liu, K. Y. Zhang, S. P.-Y. Li and T. S.-M. Tang, Chem. Commun., 2013, 49, 4271–4273 RSC.
- J. Ohata, F. Vohidov, A. Aliyan, K. Huang, A. A. Marti and Z. T. Ball, Chem. Commun., 2015, 51, 15192–15195 RSC.
- L. C.-C. Lee, J. C.-W. Lau, H.-W. Liu and K. K.-W. Lo, Angew. Chem., 2016, 128, 1058–1061 CrossRef.
- S. P.-Y. Li, A. M.-H. Yip, H.-W. Liu and K. K.-W. Lo, Biomaterials, 2016, 103, 305–313 CrossRef CAS PubMed.
- L. C.-C. Lee, H. M.-H. Cheung, H.-W. Liu and K. K.-W. Lo, Chem. – Eur. J., 2018, 24, 14064–14068 CrossRef CAS PubMed.
- Q. Zhao, M. Yu, L. Shi, S. Liu, C. Li, M. Shi, Z. Zhou, C. Huang and F. Li, Organometallics, 2010, 29, 1085–1091 CrossRef CAS.
- J. Wang, J. Xue, Z. Yan, S. Zhang, J. Qiao and X. Zhang, Angew. Chem., 2017, 129, 15124–15128 CrossRef.
- S. Tobita and T. Yoshihara, Curr. Opin. Chem. Biol., 2016, 33, 39–45 CrossRef CAS PubMed.
- K. K.-W. Lo and K. Y. Zhang, RSC Adv., 2012, 2, 12069–12083 RSC.
- P. V. Laakso, R. Robinson and H. P. Vandrewala, Tetrahedron, 1957, 1, 103–118 CrossRef CAS.
- K. K.-W. Lo, Acc. Chem. Res., 2015, 48, 2985–2995 CrossRef CAS PubMed.
- F. Liu, Y. Liang and K. N. Houk, Acc. Chem. Res., 2017, 50, 2297–2308 CrossRef CAS PubMed.
- F. M. Bickelhaupt and K. N. Houk, Angew. Chem., Int. Ed., 2017, 56, 10070–10086 CrossRef CAS PubMed.
- Y. Liang, J. L. Mackey, S. A. Lopez, F. Liu and K. N. Houk, J. Am. Chem. Soc., 2012, 134, 17904–17907 CrossRef CAS PubMed.
- D. H. Ess and K. N. Houk, J. Am. Chem. Soc., 2007, 129, 10646–10647 CrossRef CAS PubMed.
- H. Tao, F. Liu, R. Zeng, Z. Shao, L. Zou, Y. Cao, J. M. Murphy, K. N. Houk and Y. Liang, Chem. Commun., 2018, 54, 5082–5085 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthetic procedures, kinetic measurements, cytotoxicity data, computational details, copies of NMR spectra and HPLC traces. See DOI: 10.1039/c9cc06828g |
| ‡ Current address: Department of Electron Microscopy & Molecular Pathology, Cyprus Institute of Neurology & Genetics, Nicosia, Cyprus. |
| This journal is © The Royal Society of Chemistry 2019 |