
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Using alcohols as simple H2-equivalents for copper-catalysed transfer semihydrogenations of alkynes†
Trinadh
Kaicharla
 ,
Birte M.
Zimmermann
,
Martin
Oestreich
and
Johannes F.
Teichert
*
,
Birte M.
Zimmermann
,
Martin
Oestreich
and
Johannes F.
Teichert
*
Institut für Chemie, Technische Universität Berlin, Strasse des 17. Juni 115, 10623 Berlin, Germany. E-mail: johannes.teichert@chem.tu-berlin.de
First published on 22nd October 2019
Abstract
Copper(I)/N-heterocyclic carbene complexes enable a transfer semihydrogenation of alkynes employing simple and readily available alcohols such as isopropanol. The practical overall protocol circumvents the use of commonly employed high pressure equipment when using dihydrogen (H2) on the one hand, and avoids the generation of stoichiometric silicon-based waste on the other hand, when hydrosilanes are used as terminal reductants.
Catalytic transfer hydrogenations (TH) are attractive for the reduction of a variety of functional groups, as they offer practical protocols circumventing the need for high pressure equipment.1 In general, for catalytic TH there is a need for easy-to-use and safe dihydrogen (H2) sources, as these reactants are associated with the generation of one molecule of waste per turnover. Alcohols, such as the commonly employed isopropanol, fulfil the abovementioned criteria.1
Copper-catalysed reductive transformations (so-called copper hydride catalysis)2 pose formidable challenges with regard to transfer hydrogenations: On the one hand, copper hydride catalysis almost exclusively relies on hydrosilanes as reducing agents,2 bringing forward a problem of atom economy due to silicon-based waste generated. On the other hand, copper(I)-catalysed homogeneous hydrogenations (based on the atom economic H2) as investigated by us and others3–5 generally require elevated H2 pressure, hampering their overall practicability. Finally, copper is a readily available metal, which renders it highly attractive for catalytic (transfer) hydrogenations.6 It comes to a surprise that for homogeneous copper hydride catalysis, transfer hydrogenations using alcohols as H2 equivalents have not been reported so far, as this would manifest an atom economic and practical access to copper hydride chemistry, addressing the abovementioned limitations.7–9 Herein, we show that simple alcohols (such as isopropanol) can be employed as H2 equivalents for a prototypical copper hydride-based catalytic transformation, namely the semireduction of internal alkynes to alkenes.10–12
The alkyne semihydrogenation poses challenges in stereoselectivity (E vs. Z) and chemoselectivity (overreduction of the formed alkene to the corresponding alkane, Scheme 1),10 and thus serves as an ideal model reaction for catalyst development. To probe a possible transfer of a hydrogen atom from the α-position of an alcohol by means of a copper(I) catalyst, we investigated the transfer semihydrogenation of internal alkyne 1, using α,α-dideuterated benzyl alcohol (2-d2, Scheme 1). Employing readily available [SIMesCuCl]13 in catalytic amounts (10 mol%) and NaOtBu as base, full conversion either with microwave or conventional heating to the corresponding alkene 3 was found. Furthermore, significant 2H-incorporation (19–58%) in the β-position of the styrene moiety of 3 was observed, which suggested the envisaged presence of a copper(I) hydride/deuteride intermediate.14–16
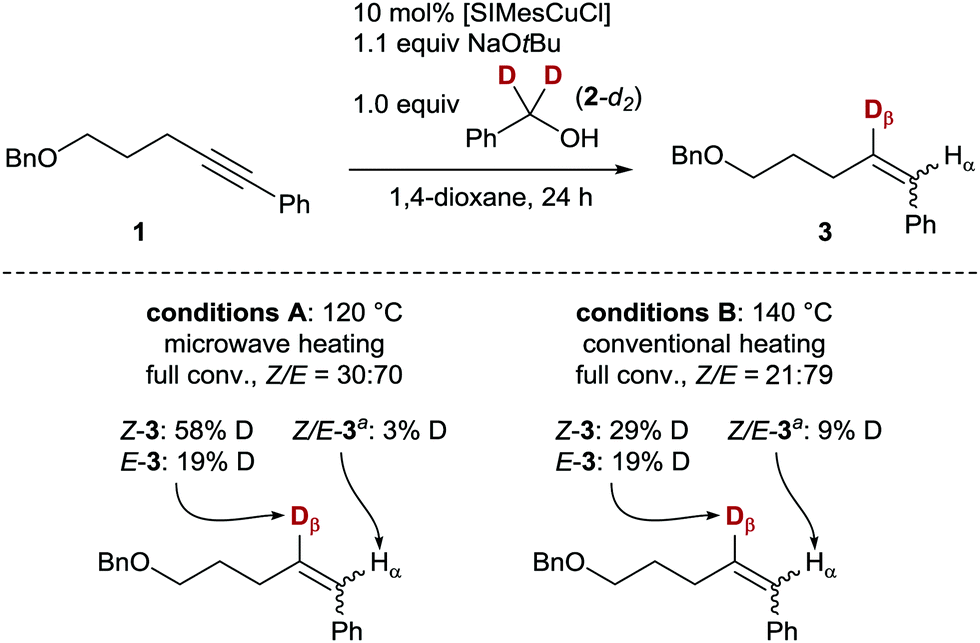 | ||
| Scheme 1 Deuterated alcohol as formal HD source in a copper-catalysed transfer hydrogenation. Reaction was carried out on a 0.2 mmol scale. Conversion monitored by GC analysis. Deuterium incorporation determined by 1H/2H NMR spectroscopy. a The Hα resonances could not be resolved by 2H NMR spectroscopy. | ||
No formation of the overreduced alkane was observed in this initial experiment, which encouraged us to further develop the copper-catalysed transfer semihydrogenation of alkynes. Therefore, the viability of alcohols as H2 sources for copper hydride catalysis in a transfer hydrogenation setting was established.
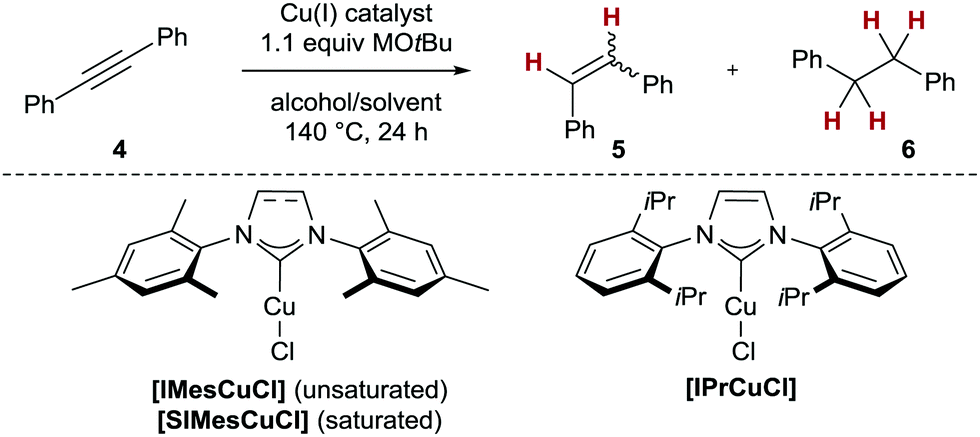
To optimise the conditions of the catalytic protocol, we opted for tolane (4) as model substrate (Table 1). We first demonstrated that 10 mol% of well-defined copper(I) complexes of standard N-heterocyclic carbene (NHC) ligands17 (SIMes, IMes and IPr, respectively)18 sufficed to achieve high or full conversion in the catalytic transfer semihydrogenation of 4 employing readily available isopropanol as H2 equivalent (Table 1, entries 1–5). Along these lines, [SIMesCuCl] as catalyst delivered the highest conversion and stereoselectivity to give stilbene Z-5 (100%, Z/E = 92![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :8, Table 1, entry 2). Notably, none of the NHC/copper(I) complexes investigated gave any overreduction to 1,2-diphenylethane (6). Other primary and/or secondary alcohols such as benzyl alcohol (2), glycerol or ethanol also gave detectable conversion, however, these H2 equivalents could not compete with isopropanol in terms of either conversion or Z/E selectivity (Table 1, entries 6–8). The change in stereoselectivity is an indication that the alcohol employed alters the structure and/or reactivity of the active catalyst. Indeed, when other secondary alcohols (2-pentanol and 2-hexanol) were tested, also, unselective reactions in terms of stereo- and chemoselectivity were observed (see the ESI,† Section S2). The omission of NHC ligands led to diminished or complete loss of chemoselectivity, as with CuCl itself either 6% (with NaOtBu as base) or complete conversion to undesired alkane 6 (using LiOtBu) was detected (Table 1, entries 9 and 10). These results indicate the presence of heterogeneous copper catalysts when no ligand is employed (see also Scheme 3 for more details). Finally, we established that under optimised conditions, the reaction could also be carried out with microwave heating to deliver the same results as conventional heating (Table 1, entry 11).
:8, Table 1, entry 2). Notably, none of the NHC/copper(I) complexes investigated gave any overreduction to 1,2-diphenylethane (6). Other primary and/or secondary alcohols such as benzyl alcohol (2), glycerol or ethanol also gave detectable conversion, however, these H2 equivalents could not compete with isopropanol in terms of either conversion or Z/E selectivity (Table 1, entries 6–8). The change in stereoselectivity is an indication that the alcohol employed alters the structure and/or reactivity of the active catalyst. Indeed, when other secondary alcohols (2-pentanol and 2-hexanol) were tested, also, unselective reactions in terms of stereo- and chemoselectivity were observed (see the ESI,† Section S2). The omission of NHC ligands led to diminished or complete loss of chemoselectivity, as with CuCl itself either 6% (with NaOtBu as base) or complete conversion to undesired alkane 6 (using LiOtBu) was detected (Table 1, entries 9 and 10). These results indicate the presence of heterogeneous copper catalysts when no ligand is employed (see also Scheme 3 for more details). Finally, we established that under optimised conditions, the reaction could also be carried out with microwave heating to deliver the same results as conventional heating (Table 1, entry 11).
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Catalyst, loading | Base | Alcohol/solventb | Conv. [%] | Z/E-5 | 6 [%] |
|
a
Reactions were carried out on a 0.2 mmol scale. Conversion and selectivity determined by GC and 1H NMR analysis.
b If not noted otherwise, 1 mL of the corresponding alcohol was employed as solvent.
c 50 mol% NaOtBu was employed.
d 2.0 equiv. BnOH in 1 mL 1,4-dioxane was used.
e 1 mL of a glycerol/1,4-dioxane mixture (1:10 v/v) was employed.
f Reaction was performed with microwave heating at 120 °C for 16 h.
|
||||||
| 1 | [SIMesCuCl], 5 mol% | NaOtBu | iPrOH | 49 | 90:10 |
<1 |
| 2 | [SIMesCuCl], 10 mol% | NaOtBu | iPrOH | 100 |
92![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :8 :8
|
<1 |
| 3 | [IPrCuCl], 10 mol% | NaOtBu | iPrOH | 90 | 92:8 |
<1 |
| 4 | [IMesCuCl], 10 mol% | NaOtBu | iPrOH | 100 | 90:10 |
<1 |
| 5 | [SIMesCuCl], 10 mol% | NaOtBuc | iPrOH | 100 | 84:16 |
<1 |
| 6 | [SIMesCuCl], 10 mol% | NaOtBu | BnOH/1,4-dioxaned | 100 | 70:30 |
<1 |
| 7 | [SIMesCuCl], 10 mol% | NaOtBu | Glycerol/1,4-dioxanee | 69 | 93:7 |
<1 |
| 8 | [SIMesCuCl], 10 mol% | NaOtBu | EtOH | 29 | 86:14 |
<1 |
| 9 | CuCl, 20 mol% | NaOtBu | iPrOH | 100 | 80:20 |
6 |
| 10 | CuCl, 20 mol% | LiOtBu | iPrOH | 100 | >1% | >99 |
| 11 | [SIMesCuCl], 10 mol% | NaOtBu | iPrOH | 100 |
95:5
|
<1 |
As the conduction of the reaction outside the microwave leads to a simpler overall procedure, we investigated the substrate scope of the copper(I)-catalysed transfer semihydrogenation with conventional heating next (Scheme 2). A variety of tolane derivatives gave the corresponding products 8a–c in very good yields and Z-stereoselectivity (≥94%, Z/E ≥ 91:9). Both electron-rich as well as electron-poor stilbene derivatives 8d to 8g were obtained with similarly good results in terms of yield and Z/E-selectivity, albeit that trifluoro-substituted stilbene 8g was isolated with a somewhat lower yield of 66%. Moving on to aryl alkyl-substituted alkynes as substrates, we were able to isolate benzyl-protected hexenol-derivative 8h with Z/E = 82:18 and in 90% yield. This result is noteworthy, as no benzyl ether deprotection was detected, albeit the overall reducing conditions of the overall process.19 The sterically more demanding cyclohexyl derivative 8i was the only compound studied which was accompanied by a negligible amount of the corresponding alkane (4%). The clean isolation of cyclopropyl styrene 8j with very good results (84%, Z/E = 97:3), and no evidence for a ring-opening gives an important indication that no radical intermediates are involved in the overall process.20 Finally, the fully stereoselective production of Z-6-dodecene (Z-8k) in acceptable yields shows that isopropanol can also be used as H2 equivalent with dialkyl alkynes as substrates, which are relatively unreactive in copper-based semihydrogenations.3
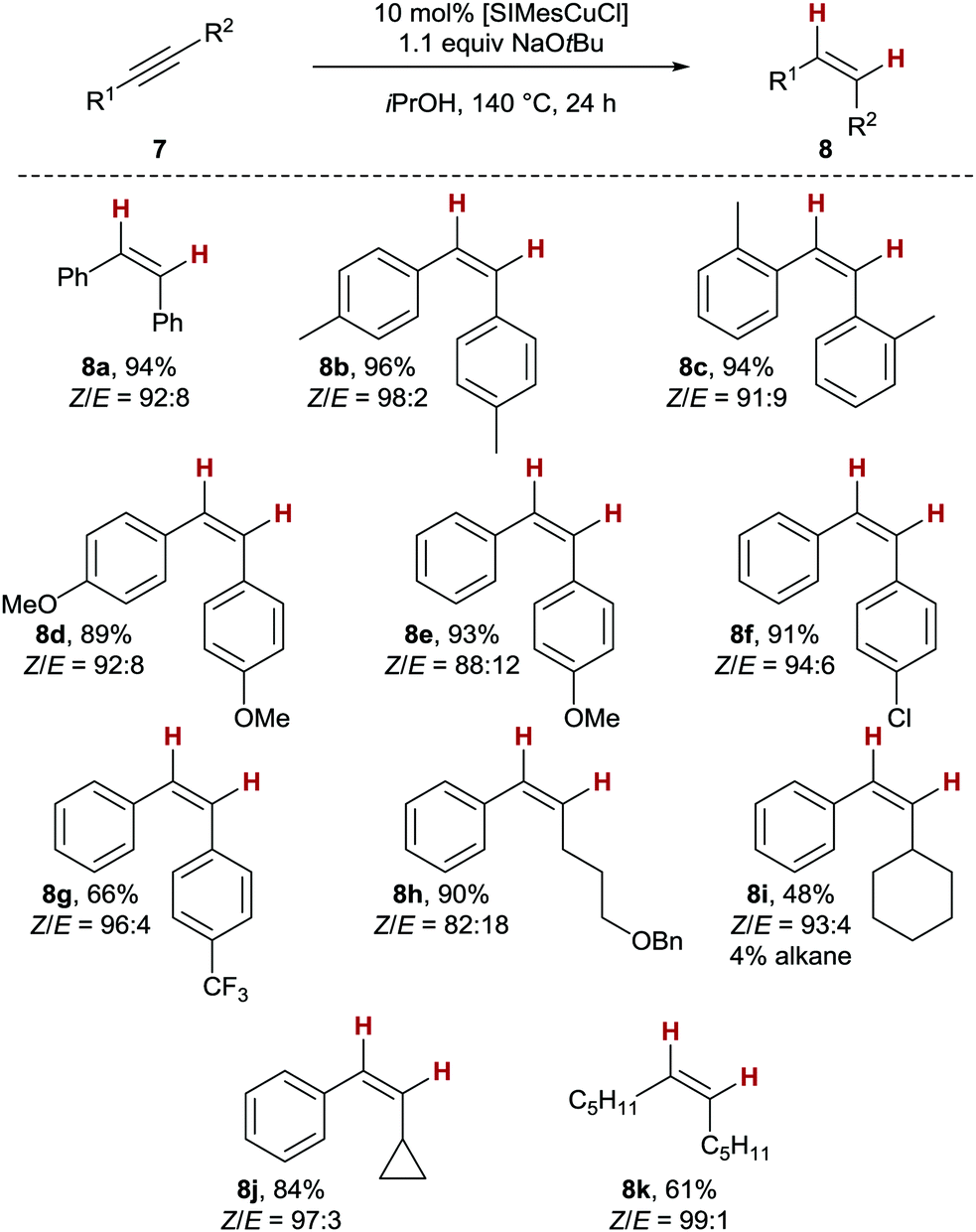 | ||
| Scheme 2 Copper-catalysed transfer semihydrogenation, substrate scope. | ||
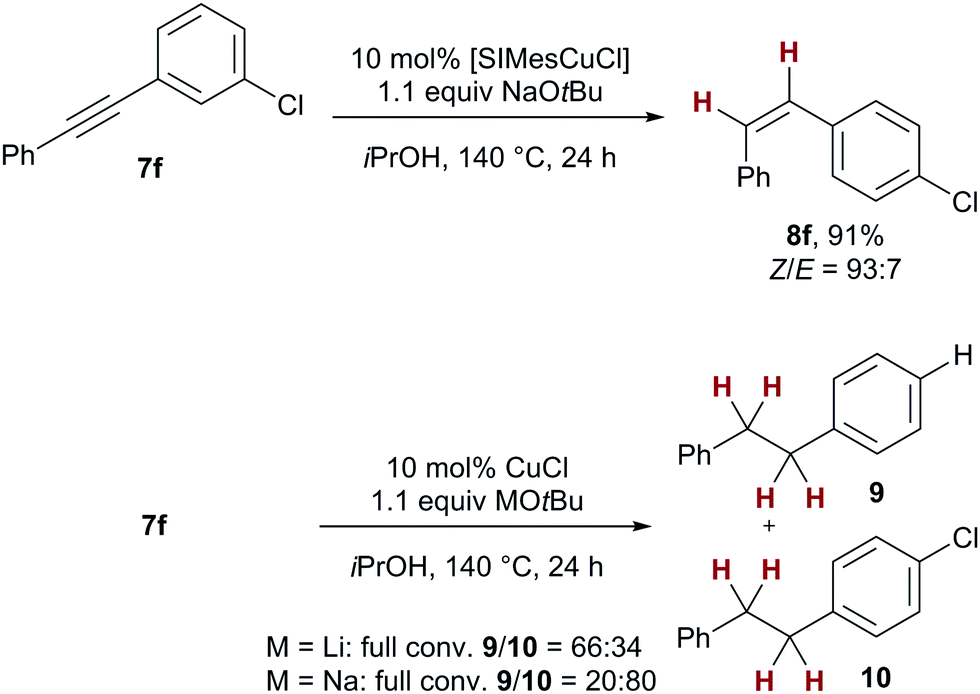 | ||
| Scheme 3 Influence of the NHC ligand on chemoselectivity. | ||
It should be noted that generally, no overreduction to the corresponding alkane was observed, underscoring the ability of the NHC ligand to control chemoselectivity in the transfer hydrogenation. This feature is in stark contrast to related transfer hydrogenations employing catalytic amounts of copper nanoparticles which cleanly deliver the corresponding alkanes from alkyne starting materials.7a The present protocol thus achieves hitherto unreached chemoselectivity for copper-catalysed transfer semihydrogenations, which can be related back to the key influence of NHC ligands. The high Z selectivity reached for most alkynes deserves some more comments: When following the reaction with 8h, a relatively fast aforegoing alkyne semihydrogenation takes place (100% conversion reached after 4h), followed by a period in which no alkane was formed but the Z/E ratio diminished, indicating a secondary isomerisation process (see the ESI† for details). These results show that close optimization of the reaction time for an alkyne of interest can lead to even higher Z/E ratios.
To underscore the significant presence of the SIMes ligand on the selectivity of the transfer semihydrogenation, we have re-investigated chlorotolane 7f as substrate. In the presence of SIMesCuCl under standard conditions, clean and stereoselective formation of the corresponding chlorostilbene 8f was observed (91%, Z/E = 93:7, see Scheme 2). Under identical reaction conditions but employing only copper(I) chloride as catalyst, no alkene formation was observed, instead high conversion to the corresponding dechlorinated and chlorinated overreduced alkane 9 and 10 was detected (9/10 = 66:34). The amount of dechlorination also depended on the counterion of the base employed: with lithium tert-butoxide, a higher percentage of the dechlorinated alkane 9 was found. (9/10 = 66:34 vs.9/10 = 20:80 with NaOtBu) This result serves as additional evidence for the fact that the active catalyst present in the transfer semihydrogenations comprises an NHC ligand which is vital for high selectivities.21
To address the need for readily available and cheap H2 equivalents for transfer hydrogenations, we have demonstrated that simple alcohols can be employed in copper-catalysed transfer hydrogenations. The results show that in the realm of copper hydride catalysis both, high pressure equipment (when using H2) as well as waste-generating hydrosilanes can be circumvented, offering a cheap access to copper hydrides and a practical overall protocol. The use of NHCs as key ligands leads to high chemoselectivity (little or no overreduction to the corresponding alkanes) and stereoselectivity (very high Z/E ratios observed). The present first entry way to copper hydride catalysis in a transfer hydrogenation setting from alcohols thus has implications for a potential broader application in reductive copper-catalysed transformations. The extension of this methodology to other common copper hydride-based transformations is currently ongoing.
This work was supported by the German Research Council (DFG, Emmy Noether Fellowship for J. F. T., TE1101/2-1; funding for T. K. through the cluster of excellence Unifying Concepts in Catalysis).
Conflicts of interest
There are no conflicts to declare.Notes and references
- For reviews on catalytic transfer hydrogenations, see: (a) D. Wang and D. Astruc, Chem. Rev., 2015, 115, 6621–6686 CrossRef CAS PubMed; (b) T. Ikariya and A. J. Blacker, Acc. Chem. Res., 2007, 40, 1300–1308 CrossRef CAS PubMed; (c) S. Gladiali and E. Alberico, Chem. Soc. Rev., 2006, 35, 226–236 RSC.
- For reviews on copper hydride catalysis, see: (a) A. J. Jordan, G. Lalic and J. P. Sadighi, Chem. Rev., 2016, 116, 8318–8372 CrossRef CAS PubMed; (b) C. Deutsch and N. Krause, Chem. Rev., 2008, 108, 2916–2927 CrossRef CAS PubMed; (c) S. Rendler and M. Oestreich, Angew. Chem., Int. Ed., 2007, 46, 498–504 CrossRef CAS PubMed.
- For copper(I)-catalysed alkyne semihydrogenations, see: (a) F. Pape, N. O. Thiel and J. F. Teichert, Chem. – Eur. J., 2015, 21, 15934–15938 CrossRef CAS PubMed; (b) K. Semba, R. Kameyama and Y. Nakao, Synlett, 2015, 318–322 CrossRef CAS; (c) N. O. Thiel and J. F. Teichert, Org. Biomol. Chem., 2016, 14, 10660–10666 RSC; (d) T. Wakamatsu, K. Nagao, H. Ohmiya and M. Sawamura, Organometallics, 2016, 35, 1354–1357 CrossRef CAS; (e) N. O. Thiel, S. Kemper and J. F. Teichert, Tetrahedron, 2017, 73, 5023–5028 CrossRef CAS; (f) F. Pape and J. F. Teichert, Synthesis, 2017, 2470–2482 CAS.
- For the related copper(I)-catalysed alkyne semireductions employing hydrosilanes, see: (a) K. Semba, T. Fujihara, T. Xu, J. Terao and Y. Tsuji, Adv. Synth. Catal., 2012, 354, 1542–1550 CrossRef CAS; (b) A. M. Whittaker and G. Lalic, Org. Lett., 2013, 15, 1112–1115 CrossRef CAS PubMed.
- For the related copper(I)-catalysed transfer semihydrogenation of alkynes with other reducing agents, see: (a) H. Cao, T. Chen, Y. Zhou, D. Han, S.-F. Yin and L.-B. Han, Adv. Synth. Catal., 2014, 356, 765–769 CrossRef CAS; (b) Ref. 12.
- For reviews on catalytic hydrogenations with abundant metals, see for example: (a) T. N. Gieshoff and A. J. von Wangelin, in Non-noble metal catalysis, Molecular approaches and reactions, ed. R. J. M. Gebbink and M.-E. Moret, Wiley-VCH, Weinheim, 2019, vol. 39, pp. 97–126 Search PubMed; (b) F. Kallmeier and R. Kempe, Angew. Chem., Int. Ed., 2018, 57, 46–60 CrossRef CAS PubMed; (c) G. A. Filonenko, R. van Putten, E. J. M. Hensen and E. A. Pidko, Chem. Soc. Rev., 2018, 47, 1459–1483 RSC; (d) P. J. Chirik, Acc. Chem. Res., 2015, 48, 1687–1695 CrossRef CAS PubMed; (e) R. M. Bullock, Science, 2013, 342, 1054–1055 CrossRef CAS PubMed.
- For the related transfer hydrogenations of alkynes with heterogeneous copper catalysts employing alcohols as H2 equivalents, see: (a) T. Subramanian and K. Pitchumani, Catal. Sci. Technol., 2012, 2, 296–300 RSC; (b) H. Bao, B. Zhou, H. Jin and Y. Liu, J. Org. Chem., 2019, 84, 3579–3589 CrossRef CAS PubMed.
- For the related transfer hydrogenations of carbonyl groups with heterogeneous copper catalysts employing alcohols as H2 equivalents, see: (a) K. Yoshida, C. Gonzalez-Arellano, R. Luque and P. L. Gai, Appl. Catal., A, 2010, 379, 38–44 CrossRef CAS; (b) M.-J. Zhang, D.-W. Tan, H.-X. Li, D. J. Young, H.-F. Wang, H.-Y. Li and J.-P. Lang, J. Org. Chem., 2018, 83, 1204–1215 CrossRef CAS PubMed.
- For a review on alkyne semihydrogenation with heterogeneous catalysts, see: J. A. Delgado, O. Benkirane, C. Claver, D. Curulla-Ferré and C. Godard, Dalton Trans., 2017, 46, 12381–12403 RSC.
- For reviews on alkyne semihydrogenation, see: (a) J. Lei, L. Su, K. Zeng, T. Chen, R. Qiu, Y. Zhou, C.-T. Au and S.-F. Yin, Chem. Eng. Sci., 2017, 171, 404–425 CrossRef CAS; (b) C. Oger, L. Balas, T. Durand and J.-M. Galano, Chem. Rev., 2013, 113, 1313–1350 CrossRef CAS PubMed; (c) M. Crespo-Quesada, F. Cárdenas-Lizana, A.-L. Dessimoz and L. Kiwi-Minsker, ACS Catal., 2012, 2, 1773–1786 CrossRef CAS; (d) A. M. Kluwer and C. J. Elsevier, in The handbook of homogeneous hydrogenation, ed. J. G. de Vries and C. J. Elsevier, Wiley-VCH, Weinheim, 2008, pp. 374–411 Search PubMed; (e) Á. Molnár, A. Sárkány and M. Varga, J. Mol. Catal. A: Chem., 2001, 173, 185–221 CrossRef.
- For selected examples of stereoselective alkyne semihydrogenations with other metals, see: (a) A. Bacchi, M. Carcelli, M. Costa, A. Leporati, E. Leporati, P. Pelagatti, C. Pelizzi and G. Pelizzi, J. Organomet. Chem., 1997, 535, 107–120 CrossRef CAS; (b) C. Belger, N. M. Neisius and B. Plietker, Chem. – Eur. J., 2010, 16, 12214–12220 CrossRef CAS PubMed; (c) M. W. van Laren and C. J. Elsevier, Angew. Chem., Int. Ed., 1999, 38, 3715–3717 CrossRef CAS.
- For a example of copper(I)-catalysed transfer hydrogenation with ammonia borane, see: E. Korytiákova, N. O. Thiel, F. Pape and J. F. Teichert, Chem. Commun., 2017, 53, 732–735 RSC.
- (SIMes = 1,3-Bis(2,4,6-trimethylphenyl)-4,5-dihydroimidazol-2-ylidene). For reviews on copper/NHC complexes, see: (a) F. Lazreg, F. Nahra and C. S. J. Cazin, Coord. Chem. Rev., 2015, 293-294, 48–79 CrossRef CAS; (b) J. D. Egbert, C. S. J. Cazin and S. P. Nolan, Catal. Sci. Technol., 2013, 3, 912–926 RSC.
- (a) N. P. Mankad, D. S. Laitar and J. P. Sadighi, Organometallics, 2004, 23, 3369–3371 CrossRef CAS; (b) A. J. Jordan, C. M. Wyss, J. Bacsa and J. P. Sadighi, Organometallics, 2016, 35, 613–616 CrossRef CAS.
- Circumstantial evidence exists for the presence of benzaldehyde-d1 after the reaction occurred. In other reactions employing 2 as H2 equivalent, deuterated benzaldehyde could be detected by 2H NMR spectroscopy. See the ESI† for details.
- No significant induction period for either heating method (A or B) was observed (∼30% conversion of 1 after 30 min), indicating a homogeneous catalyst (see the ESI† for more details). Over the course of time, the ratio of Z-3/E-3 diminished, suggesting a rapid Z-selective semihydrogenation followed by a Z → E isomerisation process. Literature precedence exists for these combined mechanisms, see for example: (a) D. Srimani, Y. Diskin-Posner, Y. Ben-David and D. Milstein, Angew. Chem., Int. Ed., 2013, 52, 14131–14134 CrossRef CAS; (b) K. Tokmic and A. R. Fout, J. Am. Chem. Soc., 2016, 138, 13700–13705 CrossRef CAS PubMed; (c) K. Murugesan, C. B. Bheeter, P. R. Linnebank, A. Spannenberg, J. N. H. Reek, R. V. Jagadeesh and M. Beller, ChemSusChem, 2019, 12, 3363–3369 CrossRef CAS PubMed.
- For selected overviews on NHC ligands, see: (a) S. P. Nolan, N-Heterocyclic carbenes, Effective tools for organometallic synthesis, Wiley-VCH, Weinheim, 2014 Search PubMed; (b) M. N. Hopkinson, C. Richter, M. Schedler and F. Glorius, Nature, 2014, 510, 485–496 CrossRef CAS PubMed; (c) J. C. Y. Lin, R. T. W. Huang, C. S. Lee, A. Bhattacharyya, W. S. Hwang and I. J. B. Lin, Chem. Rev., 2009, 109, 3561–3598 CrossRef CAS PubMed; (d) V. Cesar, S. Bellemin-Laponnaz and L. H. Gade, Chem. Soc. Rev., 2004, 33, 619–636 RSC.
- (IMes = 1, 3-Bis(2,4,6-trimethylphenyl)-imidazol-2-ylidene, IPr = 1,3-Bis(2,6-di-iso-propylphenyl)-imidazol-2-ylidene.
- Generally, reductive/hydrogenative conditions are employed for the cleavage of benzyl ethers, see: (a) P. G. M. Wuts and T. W. Greene, Greene's protective groups in organic synthesis, Wiley, Hoboken, 4th edn, 2007 Search PubMed; (b) P. J. Kocieński, Protecting groups, Thieme, Stuttgart, 3rd edn, 2004 Search PubMed.
- (a) M. Newcomb, Tetrahedron, 1993, 49, 1151–1176 CrossRef CAS; (b) D. Griller and K. U. Ingold, Acc. Chem. Res., 1980, 13, 317–323 CrossRef CAS.
- In the related heterogeneous copper-catalysed hydrogenations, NHC ligands have also been shown to significantly improve stereo- and chemoselectivity, see: (a) A. Fedorov, H.-J. Liu, H.-K. Lo and C. Copéret, J. Am. Chem. Soc., 2016, 138, 16502–16507 CrossRef CAS PubMed; (b) N. Kaeffer, H.-J. Liu, H.-K. Lo, A. Fedorov and C. Copéret, Chem. Sci., 2018, 9, 5366–5371 RSC; (c) N. Kaeffer, K. Larmier, A. Fedorov and C. Copéret, J. Catal., 2018, 364, 437–445 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, characterisation and NMR spectra. See DOI: 10.1039/c9cc06637c |
| This journal is © The Royal Society of Chemistry 2019 |