
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Monitoring fast chemical processes by reaction-interrupted excitation transfer (ExTra) NMR spectroscopy†
Gabriel E.
Wagner‡
a,
Sebastian
Tassoti‡
 b,
Simon
Glanzer
b,
Eduard
Stadler
c,
Rainer
Herges
d,
Georg
Gescheidt
c and
Klaus
Zangger
*b
b,
Simon
Glanzer
b,
Eduard
Stadler
c,
Rainer
Herges
d,
Georg
Gescheidt
c and
Klaus
Zangger
*b
aInstitute of Hygiene, Microbiology and Environmental Medicine, Medical University of Graz, Neue Stiftingtalstraße 6, A 8010 Graz, Austria
bInstitute of Chemistry, Organic and Bioorganic Chemistry, University of Graz, Heinrichstraße 28, A-8010 Graz, Austria. E-mail: klaus.zangger@uni-graz.at
cInstitute of Physical and Theoretical Chemistry, Graz University of Technology, Stremayrgasse 9, A-8010 Graz, Austria
dOtto Diels Institute for Organic Chemistry, University of Kiel, Otto-Hahn-Platz 4, DE-24118 Kiel, Germany
First published on 26th September 2019
Abstract
NMR spectroscopy is generally used to investigate molecules under equilibrium conditions. Despite recent technological and methodogical developments to study on-going reactions, tracing the fate of individual atoms during an irreversible chemical reaction is still a challenging and elaborate task. Reaction-interrupted excitation transfer (ExTra) NMR provides a selective tracking of resonances from atoms, which undergo chemical conversion. We show that reactions triggered either by rapid mixing or by photo-excitation can be conveniently followed at a sub-second time scale using standard NMR equipment. In ExTra NMR we use the selectively inverted magnetization of a selected atom to follow its conversion in the course of a fast chemical reaction. The chemical reaction has to be started within the relaxation period of an initial inverting 180° pulse. The presented protocol provides a generally applicable NMR method for reaction monitoring.
NMR spectroscopy is a powerful, widely used tool for the investigation of chemical reactions. It has been used to study kinetics of unstable reaction intermediates, mechanisms in organic chemistry1,2 and for process control in inline/online NMR.3,4 Early on it has been extended to the field of biochemistry, where it is an important tool for the investigation of enzyme reactions, protein folding,5,6 or more recently for the study of fast kinetics of pH-induced protein unfolding.7 These versatile fields of application result from the extensive structural information that can be derived from the chemical shift and multiplet structures of the spectra.8–10 A further advantage compared to other methods is that there is no necessity for a prior calibration, as the integral of a signal is directly proportional to the quantity of the material present.11
NMR is routinely used to study molecular rearrangements of fast reversible reactions by EXSY type experiments,12,13 where recently an experiment for the monitoring of photochemical equilibria was proposed (PC-EXSY).14 Similar to these methods are SCOTCH type experiments, where magnetisation is transferred during the mixing time of a modified NOESY pulse sequence.15,16 For other photo-induced reactions, photo-CIDNP is particularly useful and provides a simple tool to study the interactions of proteins with cofactors, inhibitors and other molecules of choice.17
The method of choice for slow reversible and irreversible reactions is usually the acquisition of a series of spectra. For instance, ultrafast 2D NMR is used for the real-time monitoring of redox reactions.18 One of the drawbacks in the latter case is the low temporal resolution of NMR spectroscopic approaches, with sampling rates usually in the range of seconds. This is due to a rather long relaxation delay, which is needed to avoid successive signal saturation. However, several methods have been proposed to circumvent this problem, including the addition of paramagnetic relaxation agents,19 to lower relaxation times20–23 or the use of smaller pulse-angles and purge gradients.24–26
For fast reactions below the microsecond scale, time-resolved CIDNP is the method of choice.27,28 In contrast, fast and irreversible reactions are difficult to study by NMR. Such reactions might already be finished before the sample can be transferred into the NMR magnet. Rapid injection devices1,29–31 or stopped flow techniques5,32 can be used to cope with this limitation. In combination with our recently published NMR experiment, which allows for a continuous excitation and acquisition of spectra without any interscan delay, this setup allows the investigation of on-going reactions at an unprecedented temporal resolution.7 Furthermore, field inhomogeneities caused by the injection of reactants and volume changes are drastically reduced because each signal is recorded in a narrow z-slice only.11
With a few exceptions, the focus of reaction monitoring by NMR spectroscopy has been on the properties shown by reactants over the course of a reaction as described above. However, if the fate of a specific atom is of interest, options are pretty limited. In general, such an issue will be addressed by the incorporation of labelled isotopes, which often is not only a cost-but also a time-intensive task. More recently, hyperpolarized NMR spectroscopy was used to achieve mechanistic insight into organic and bioorganic chemical reactions.33–38 Amongst others, Hilty and colleagues developed an experiment, where the selective inversion of a single carbon atom could be traced from the reactant to its final position within the product molecule at natural abundance by using ex situ dynamic nuclear polarization (DNP). This is possible because of the preservation of spin states even during on-going reactions and the signal increase due to DNP by a factor of roughly 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000.39–41
000.39–41
Within this work we want to extend the application of NMR-assisted tracking of individual atoms during fast reactions. Our method is specifically suited for irreversible reactions with a high conversion percentage within the first few seconds of the reaction. A reaction-interrupted pulse sequence (Fig. 1) is proposed where the inversion of the resonance of interest occurs before the injection of the reactant and not afterwards.39 So as a first step, selective inversion of one signal takes place. Then, the chemical reaction is started by rapid injection of a reactant or, for photochemical reactions, by illumination of the sample. After a sufficient reaction delay, the inverted polarization can be detected on the product. While our experiment has inherently a lower sensitivity than DNP-based techniques, it has other advantages: (I) it does not require any additional dedicated hardware and therefore it can be carried out in any lab, equipped with a standard NMR system (II) it is independent of special solvents and radicals for DNP polarization, which might interfere with the investigated reaction.
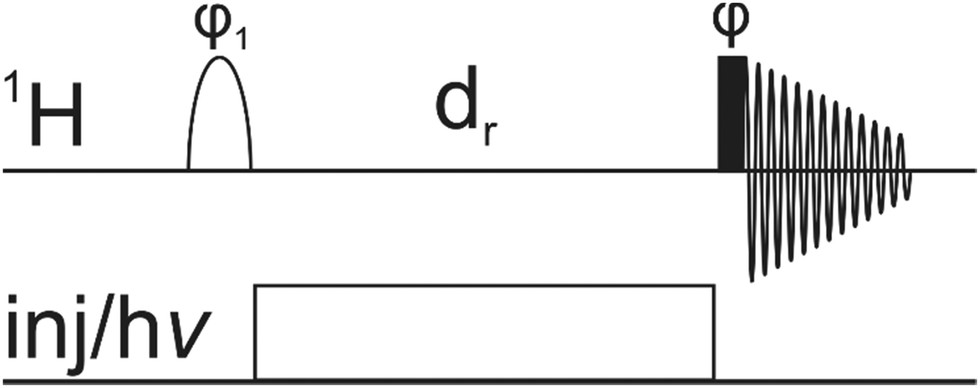 | ||
| Fig. 1 Pulse-injection/light sequence for the proposed excitation transfer reaction monitoring experiment. The resonance of interest is selectively inverted by a 180° shaped pulse. The reaction takes place during the delay dr and is triggered either by rapid injection or illumination (for photochemical reactions). The readout is achieved by a non-selective 90° hard pulse. | ||
As a proof of concept, we chose to follow the fate of the allyl alcohol's H1-proton during a bromination using the ExTra NMR approach (Fig. 2). A selective 180° Gaussian pulse was used to invert the corresponding resonance at 6.04 ppm right before the manual injection of bromine (Fig. 2a). Mixing of the reaction compounds within a dead time of roughly 100 ms right inside the NMR magnet was achieved by using a flat bottom 5 mm Shigemi tube and a rapid injection device as described by Mok et al.31 The reaction is finished almost within a second and hence allowed us to observe the transfer of the selectively inverted magnetization from the reactant to the product. The final spectrum (Fig. 2b) was recorded 1.5 seconds after the inversion pulse to allow the reaction to proceed. In the product spectrum, there is a loss in resolution, likely caused by perturbation of the sample heterogeneity by mixing. However, the fate of the mentioned proton can easily be followed due to the appearance of a negative peak in the obtained NMR spectrum of the product. This peak belongs to the H1′-proton in 2,3-dibromo-1-propanol and hence demonstrates the potential of the proposed NMR experiment.
 | ||
| Fig. 2 Depiction of the reaction scheme of the bromination of allyl alcohol and the corresponding spectra of the reactant and the product 2,3-dibromo-1-propanol: (a) conventional 1D spectrum of allyl alcohol. The resonance of the proton that is selectively inverted by an 180° Gaussian pulse during the ExTra NMR approach is marked in red. (b) The ExTra NMR sequence allows an easy identification of the previously inverted H1-proton in the product spectrum due to the appearance of a negative signal at the respective frequency (H1′-proton marked in blue). Parameters of spectra (a) and (b): 1 scan, 16k data points, 0.3 Hz exponential window function. | ||
The approach is not limited to chemically-induced reactions but can be initiated by any other trigger. An example is the isomerization of the (Z)-isomer of 5,6-dihydrodibenzo[c,g][1,2]diazocine (1) to (E)-1 by irradiation with UV-light. This reaction is fast at room temperature, producing yields of five percent and higher of (E)-1 after one second of illumination and reaching an equilibrium of both isomers after prolonged illumination (photostationary state). Green light (λ = 523 nm) can be used to switch back to the pure (Z)-isomer within 60 seconds. Switching the LED used for in situ illumination eliminates the need to remove the sample from the magnet. Therefore, the elucidation of the reaction path of one proton only takes slightly over a minute. Application of the proposed pulse sequence allows for quick and facile assignment of a selected signal of (Z)-1 to the corresponding signal of (E)-1 after a single scan. Repetition of the experiment to determine the fate of other protons is swiftly accomplished as shown in Fig. 3b–d.
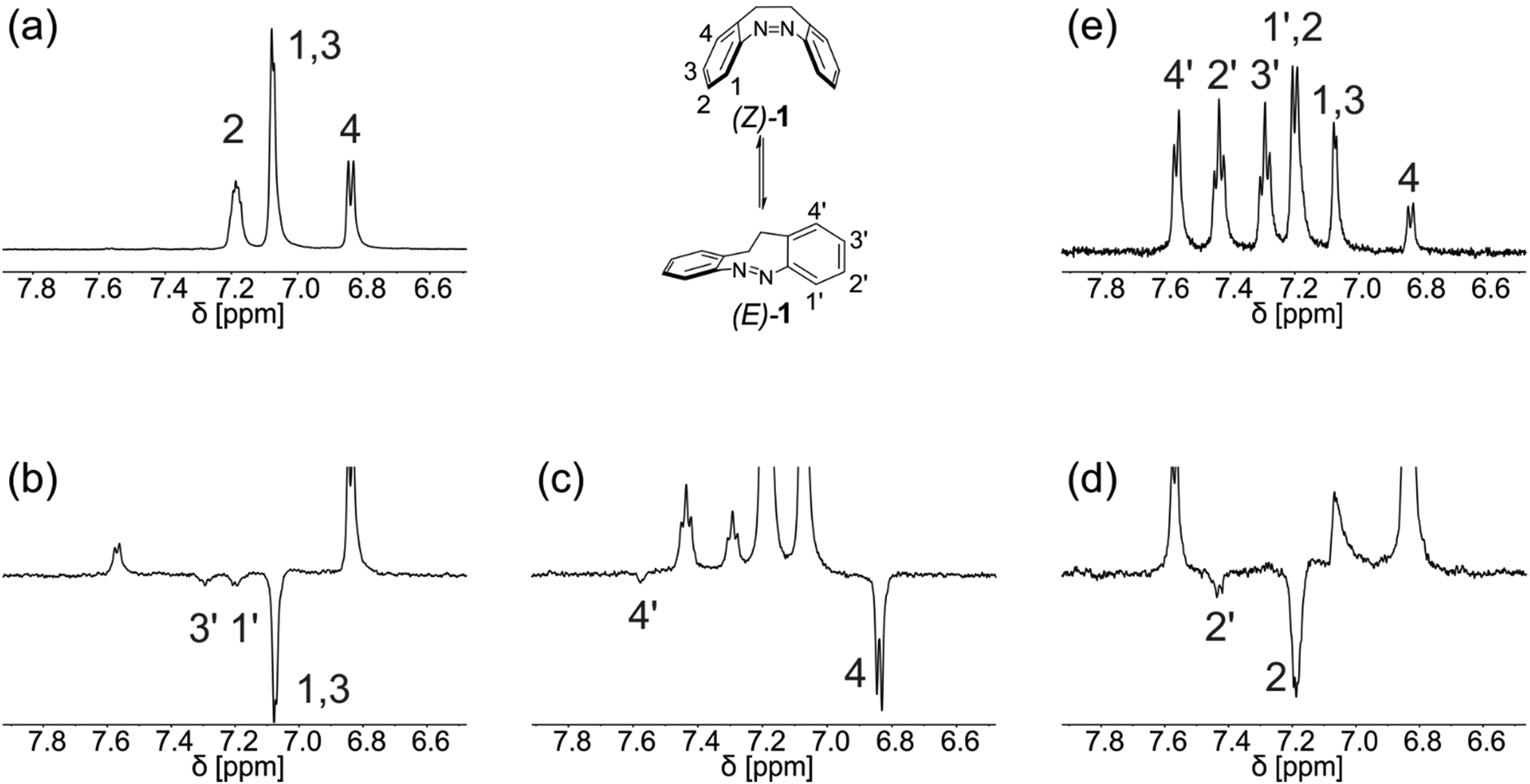 | ||
| Fig. 3 (a) Proton spectrum of the aromatic region of (Z)-1 in acetonitrile-d3 (16 scans, 64k data points, 0.3 Hz exponential window function). (b–d) Selective inversion of signals 1–4 by application of the proposed pulse-and-light sequence, allowing the correlation of each of the aromatic protons of (Z)-1 to the corresponding protons in (E)-1. (e) Assignment of all aromatic signals in the proton spectrum (1 scan, 16k data points, 0.3 Hz exponential window function) of the reaction mixture (taken after 30 s of illumination). | ||
Noteworthy, the magnetization of a certain resonance does not have to be negative in order to be traced. The conversion of the selected atom from the educts to the product is traceable as long as the reaction is triggered before the inverted magnetization relaxes: the differing intensities of product peaks in the ExTra NMR spectrum and product peaks from a normal proton spectrum recorded after the reaction clearly reveal the chemical conversion (see Fig. S1, ESI†). In place of inversion, the resonance frequency can also be saturated prior to the reaction. However, we have found that inversion gives more convincing and convenient results (see Fig. S2, ESI†).
In conclusion, we have shown that irreversible chemical reactions, which can be triggered by mixing or illumination and which proceed at rates in the millisecond time regime can be conveniently followed by NMR. A simple 1D experiment consisting of a selective 180° preparation pulse, followed by triggering the reaction and a readout 90° hard pulse provides the course of the reaction mirrored by the resonances of selected protons. Our pulse sequence has the potential of assessing a wide range of chemical reactions including protonation, complexation, halogenation and photoreactions. Limitations are the presence of NMR active nuclei participating in the reaction and being present at an appropriately high concentration. The conversion of observable amounts of product has to be faster than the relaxation of the inverted magnetisation.
Furthermore, the proposed approach can be used to get insight into reaction mechanisms, without the necessity of investigating compounds with fully assigned NMR spectra.
Our strategy allows analysing chemical conversions with standard NMR equipment (see ESI† for pulse sequences) without the requirement of isotopic labelling or the use of very specific and chemically limited DNP-based (dynamic nuclear polarization) methods.
All NMR experiments were performed on a Bruker Avance III 500 MHz spectrometer equipped with a 5 mm TXI probe with z-axis gradients at 300 K except for the photoisomerization. The spectra of the latter were recorded on a Bruker Avance III 700 MHz spectrometer using a 5 mm cryogenically cooled TCI probe with z-axis gradients.
All chemicals were purchased from Sigma Aldrich in the highest grade available and used as received. The photo-switchable molecule 5,6-dihydrodibenzo[c,g][1,2]diazocine (1) was synthesized according to published procedures.42,43
For in situ sample irradiation, light emitted from high power LEDs (lumitronix) was directly coupled into the blunt and polished end of a 4 mm thick optical fiber (ceramoptec). At the tip of the fiber, the nylon jacket was removed and a pencil shaped tip was created by sandblasting. The high-power LEDs were powered by a custom-made power source allowing for precise modulation of intensity by pulse width modulation (Sahlmann Photochemical Solutions) and were triggered by the NMR console via the IPSO-unit.
Financial support to K. Z. from the Austrian Science Foundation (FWF) (project number P-30230) and through the Doktoratskolleg Molecular Enzymology (W901) is gratefully acknowledged. We also thank the interuniversity program in natural sciences, NAWI Graz, for financial support. We would also like to acknowledge P. J. Hore and C. Jones from the Department of Chemistry at the Oxford University for supplying us with construction plans of their rapid injection device. R. H. is grateful for support by the DFG (SFB677).
Conflicts of interest
There are no conflicts to declare.Notes and references
- J. F. McGarrity and J. Prodolliet, J. Org. Chem., 1984, 49, 4465–4470 CrossRef CAS.
- S. E. Denmark, B. J. Williams, B. M. Eklov, S. M. Pham and G. L. Beutner, J. Org. Chem., 2010, 75, 5558–5572 CrossRef CAS PubMed.
- A. Brächer, L. M. Kreußer, S. Qamar, A. Seidel-Morgenstern and E. von Harbou, Chem. Eng. J., 2018, 336, 518–530 CrossRef.
- D. A. Foley, J. Wang, B. Maranzano, M. T. Zell, B. L. Marquez, Y. Xiang and G. L. Reid, Anal. Chem., 2013, 85, 8928–8932 CrossRef CAS PubMed.
- P. J. Hore, S. L. Winder, C. H. Roberts and C. M. Dobson, J. Am. Chem. Soc., 1997, 119, 5049–5050 CrossRef CAS.
- C. M. Dobson and P. J. Hore, Nat. Struct. Biol., 2002, 5, 504–507 CrossRef PubMed.
- G. E. Wagner, P. Sakhaii, W. Bermel and K. Zangger, Chem. Commun., 2013, 49, 3155 RSC.
- B. Böttcher, V. Schmidts, J. A. Raskatov and C. M. Thiele, Angew. Chem., Int. Ed., 2010, 49, 205–209 CrossRef PubMed.
- F. Von Rekowski, C. Koch and R. M. Gschwind, J. Am. Chem. Soc., 2014, 136, 11389–11395 CrossRef CAS PubMed.
- G. C. Lloyd-Jones and N. P. Taylor, Chem. – Eur. J., 2015, 21, 5423–5428 CrossRef CAS PubMed.
- J. Kind and C. M. Thiele, J. Magn. Reson., 2015, 260, 109–115 CrossRef CAS PubMed.
- B. H. Meier and R. R. Ernst, J. Am. Chem. Soc., 1979, 101, 6441–6442 CrossRef CAS.
- C. L. Perrin and T. J. Dwyer, Chem. Rev., 1990, 90, 935–967 CrossRef CAS.
- E. Stadler, S. Tassoti, P. Lentes, R. Herges, T. Glasnov, K. Zangger and G. Gescheidt, Anal. Chem., 2019, 91, 11367–11373 CrossRef CAS PubMed.
- J. Kemmink, G. W. Vuister, R. Boelens, K. Dijkstra and R. Kaptein, J. Am. Chem. Soc., 1986, 108, 5631–5633 CrossRef CAS.
- P. J. W. Pouwels and R. Kaptein, J. Phys. Chem., 1993, 97, 13318–13325 CrossRef CAS.
- J. Hore and R. W. Broadhurst, Prog. Nucl. Magn. Reson. Spectrosc., 1993, 25, 345–402 CrossRef.
- R. Boisseau, U. Bussy, P. Giraudeau and M. Boujtita, Anal. Chem., 2015, 87, 372–375 CrossRef CAS PubMed.
- E. Stadler, M. Dommaschk, P. Frühwirt, R. Herges and G. Gescheidt, ChemPhysChem, 2018, 19, 571–574 CrossRef CAS PubMed.
- N. P. Wickramasinghe, M. Kotecha, A. Samoson, J. Past and Y. Ishii, J. Magn. Reson., 2007, 184, 350–356 CrossRef CAS PubMed.
- G. Pintacuda and G. Otting, J. Am. Chem. Soc., 2002, 124, 372–373 CrossRef CAS PubMed.
- T. Madl, W. Bermel and K. Zangger, Angew. Chem., Int. Ed., 2009, 48, 8259–8262 CrossRef CAS PubMed.
- D. J. Cookson and B. E. Smith, J. Magn. Reson., 1984, 57, 355–368 CAS.
- A. Bax, T. Mehlkopf, J. Smidt and R. Freeman, J. Magn. Reson., 1980, 41, 502–506 CAS.
- R. R. Ernst, G. Bodenhausen, A. Wokaun and A. G. Redfield, Phys. Today, 1989, 42, 75–76 CrossRef.
- P. Schanda, V. Forge and B. Brutscher, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 11257–11262 CrossRef CAS PubMed.
- G. L. Closs and R. J. Miller, J. Am. Chem. Soc., 1981, 103, 3586–3588 CrossRef CAS.
- G. L. Closs, R. J. Miller and O. D. Redwine, Acc. Chem. Res., 1985, 18, 196–202 CrossRef CAS.
- S. E. Denmark, B. M. Eklov, P. J. Yao and M. D. Eastgate, J. Am. Chem. Soc., 2009, 131, 11770–11787 CrossRef CAS PubMed.
- J. F. McGarrity, J. Prodolliet and T. Smyth, Org. Magn. Reson., 1981, 17, 59–65 CrossRef CAS.
- K. H. Mok, T. Nagashima, I. J. Day, J. A. Jones, C. J. V. Jones, C. M. Dobson and P. J. Hore, J. Am. Chem. Soc., 2003, 125, 12484–12492 CrossRef CAS PubMed.
- R. O. Kühne, T. Schaffhauser, A. Wokaun and R. R. Ernst, J. Magn. Reson., 1979, 35, 39–67 Search PubMed.
- M. E. Halse, Trends Anal. Chem., 2016, 83(A), 76–83 CrossRef CAS.
- C. Hilty and S. Bowen, Org. Biomol. Chem., 2010, 8, 3361–3365 RSC.
- Y. Lee, G. S. Heo, H. Zeng, K. L. Wooley and C. Hilty, J. Am. Chem. Soc., 2013, 135, 4636–4639 CrossRef CAS PubMed.
- H. Zeng, Y. Lee and C. Hilty, Anal. Chem., 2010, 82, 8897–8902 CrossRef CAS PubMed.
- M. Ragavan, H. Y. Chen, G. Sekar and C. Hilty, Anal. Chem., 2011, 83, 6054–6059 CrossRef CAS PubMed.
- G. Zhang, F. Schilling, S. J. Glaser and C. Hilty, J. Magn. Reson., 2016, 272, 123–128 CrossRef CAS PubMed.
- S. Bowen and C. Hilty, Anal. Chem., 2009, 81, 4543–4547 CrossRef CAS PubMed.
- A. Bornet, X. Ji, D. Mammoli, B. Vuichoud, J. Milani, G. Bodenhausen and S. Jannin, Chem. – Eur. J., 2014, 20, 17113–17118 CrossRef CAS PubMed.
- X. Ji, A. Bornet, B. Vuichoud, J. Milani, D. Gajan, A. J. Rossini, L. Emsley, G. Bodenhausen and S. Jannin, Nat. Commun., 2017, 8, 13975 CrossRef CAS PubMed.
- W. W. Paudler and A. G. Zeiler, J. Org. Chem., 1969, 34, 3237–3239 CrossRef CAS.
- R. Siewertsen, H. Neumann, B. Buchheim-Stehn, R. Herges, C. Näther, F. Renth and F. Temps, J. Am. Chem. Soc., 2009, 131, 15594–15595 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9cc06427c |
| ‡ G. W. and S. T. contributed equally to the work. |
| This journal is © The Royal Society of Chemistry 2019 |