
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence“Off–on” switching of intracellular singlet oxygen release under biocompatible conditions†
Esma
Ucar
ab,
Dongmei
Xi
a,
Ozlem
Seven
c,
Cansu
Kaya
b,
Xiaojun
Peng
 a,
Wen
Sun
*a and
Engin U.
Akkaya
*a
a,
Wen
Sun
*a and
Engin U.
Akkaya
*a
aState Key Laboratory of Fine Chemicals, Department of Pharmacy, Dalian University of Technology, 2 Linggong Road, 116024, Dalian, China. E-mail: sunwen@dlut.edu.cn; eua@dlut.edu.cn
bDepartment of Chemistry, Bilkent University, 06800 Ankara, Turkey
cUNAM-National Nanotechnology Research Center, Bilkent University, 06800 Ankara, Turkey
First published on 15th October 2019
Abstract
Precise spatiotemporal control of singlet oxygen generation is of immense importance considering its involvement in photodynamic therapy. In this work, we present a rational design for an endoperoxide which is highly stable at ambient temperatures yet, can rapidly be converted into a highly labile endoperoxide, thus releasing the “stored” singlet oxygen on demand. The “off–on” chemical switching from the stable to the labile form is accomplished by the reaction with fluoride ions. The potential utility of controlled singlet oxygen release was demonstrated in cell cultures.
Fundamental limitations of the photodynamic therapy (PDT) of cancer remain in place, despite an intense effort worldwide to overcome or circumvent them.1–7 Limited penetration of light through tissues8,9 and less than optimal oxygen concentrations,10,11 especially in the most aggressive, hypoxic tumors, has kept the clinical practice of photodynamic therapy very limited. Today, the most common application12,13 of PDT, does not even make use of a proper photosensitizer, but a biological precursor of protoporphyrin-IX for topical application, followed by irradiation. More than 100 years after the initial discovery,14 the full potential of PDT remains to be fulfilled.
In our research group (Akkaya), we have been trying to address these two fundamental problems by separating photonic excitation from the actual delivery of the cytotoxic agent, i.e., singlet oxygen, in a two-stage plan for photodynamic action.15–17 This requires the development of a stable non-toxic “storage” compound, which could then be transferred to the biological target, and later, coerced to release its cargo on target (Fig. 1). The most promising class of such storage compounds are endoperoxides derived from arenes.18 These endoperoxides can be prepared ex situ, and as a result, light of any wavelength can be used for photosensitized synthesis of these endoperoxides. When successfully implemented, this approach may indeed deliver the benefits of “targeted singlet oxygen delivery” (TSOD) to tumors.
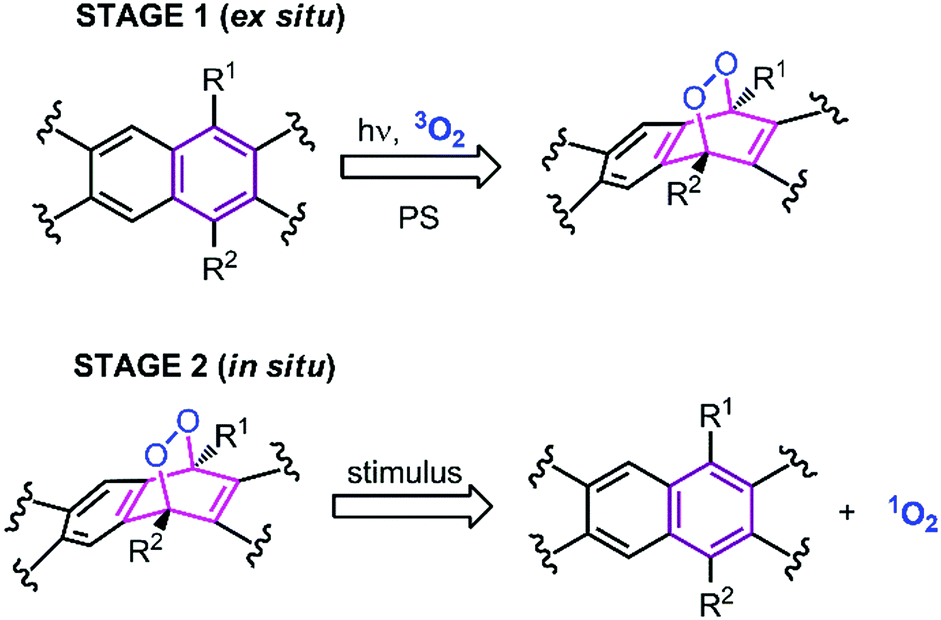 | ||
| Fig. 1 Two-stage, modified PDT concept “targeted singlet oxygen delivery or TSOD”, which does not require either O2 or light for in situ generation of singlet oxygen. R1 and R2 are typically alkyl or aryl substituents. | ||
It is possible to cause singlet oxygen release by the addition of a strong electrophile in organic solvents,19 or plasmonic heating,15 but, it is of extreme importance to find “biologically compatible” methods to control (i.e., switch “on”) the rates of singlet oxygen generation, at a minimum, a method which works in aqueous solutions, under neutral pH conditions, and at physiological temperatures.
Here, we present the first example of such fine control, together with a demonstration of its effectiveness in MCF-7 (human breast adenocarcinoma) cell cultures (cells were obtained from the Institute of Basic Medical Sciences (IBMS) of the Chinese Academy of Medical Sciences).
Thermal decomposition of 1,4-dimethylnaphthalene endo-peroxide derivatives have been studied previously (Fig. 2).20,21 Cycloreversion of the endoperoxides yields singlet oxygen as the major product, along with varying percentages of ground state (triplet) oxygen. While both steric and electronic factors influence cycloreversion rates, it seems that steric factors are predominant.
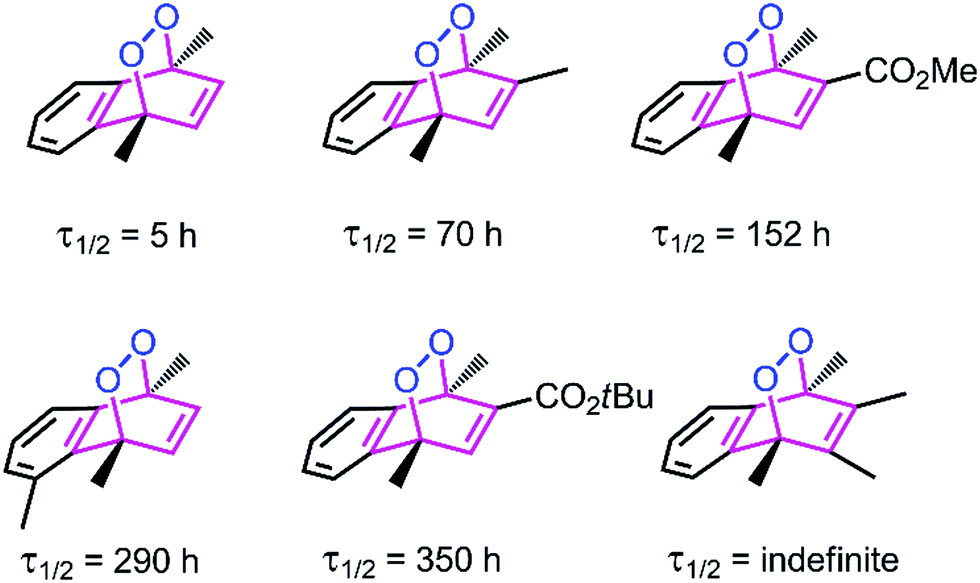 | ||
| Fig. 2 Selected literature examples20,21 of 1,4-dimethylnaphthalene endoperoxides with differing cycloreversion half-lives determined at 25 °C. Steric bulk at positions 2, 3 and 5, seems to be a significant stabilizing factor for the endoperoxide. | ||
With that understanding, we focused on reversible or removable steric barriers or stoppers for cycloreversion. The other factor to consider is the compatibility of singlet oxygen with the products. Of course, finally the removal of the steric stopper has to be carried out in aqueous solutions around pH 7.0. Among all the options, an aryl/alkyl silyl substituent (such as TMS) looked highly promising.22,23 The synthesis of endoperoxide 1 starts with 1,4-dimethylnaphthalene (ESI†). Bromination at position 2, followed by a Grignard reaction with TMS-Cl, resulted in the naphthalene derivative 3. Irradiation with a white lamp (400 W) at −20 °C, in DCM with methylene blue as photosensitizer, under an O2 atmosphere yielded the target compound 1. The endoperoxide product can easily be separated from the impurities by chromatography at ambient temperatures, in accordance with its expected stability.
We then determined the reaction rate of cycloreversion using 1H NMR spectroscopy in DMSO-d6 (Fig. 3, and ESI,† Fig. S4 and S5). Endoperoxide is highly stable at room temperature with a half-life of slightly more than 800 hours. The rate of change in 1H NMR can clearly be followed focusing on the bridgehead methyl substituents. A set of two singlets (1.75 and 1.85 ppm) diminishes, while a new set of aromatic methyl substituents at 2.60 and 2.70 ppm emerges. Then, to an identical sample, we added TBAF (tetrabutylammonium fluoride) at 20 mM. The removal of a TMS group was essentially instantaneous (Fig. 3 bottom set of spectra) and two methyl signals converge into one, as the TMS group is removed. The change is apparent even at t = 0 time point, which is just seconds after the addition of TBAF to the NMR sample. This is followed by a much faster cycloreversion of 2 to 4 with a half-life of 5 hours, in accordance with the previous literature data (Fig. 4).
 | ||
| Fig. 3 Time evolution of 1H NMR spectra of endoperoxide 1 in DMSO-d6, without added TBAF (top) and with 20 mM TBAF (bottom). Time points for the upper set of spectra are (a through g) 0, 27.5, 67.5, 115.5, 153.5, 251, 990 h. Time points for the bottom set of spectra on the right are (a through e) 0, 3.5, 20, 27.5, 45 h. Spectral data were acquired at 25 °C. | ||
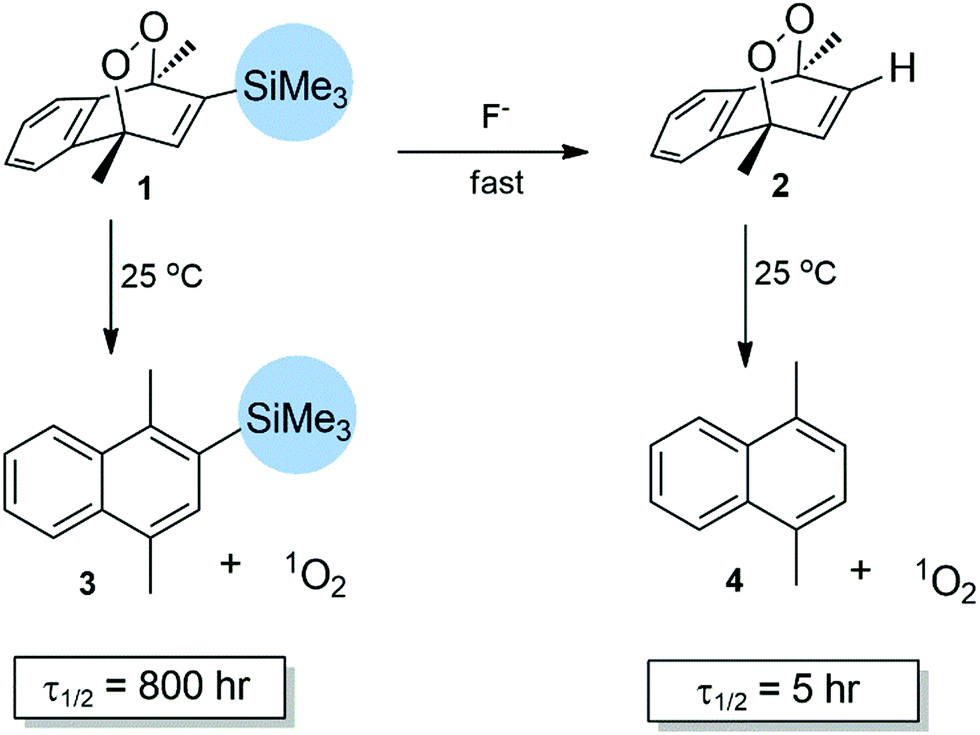 | ||
| Fig. 4 “Off–On” switching of 1O2 release: when fluoride in the form of TBAF was added in DMSO, 2-TMS-naphthalene endoperoxide 1 is rapidly converted to the parent endoperoxide 2 in a process that increases the rate of singlet oxygen release by more than two orders of magnitude. | ||
Together with the experiments using the singlet oxygen probe 1,3-diphenylisobenzofuran (DPBF) (ESI,† Fig. S6), these results provided satisfactory evidence for the fluoride mediated release of singlet oxygen from the storage compound 1. We expected that without the fluoride deprotection, the release from the endoperoxide 1 would be too slow to make any difference, especially in an aqueous medium, where the half-life of singlet oxygen is less than 3.5 microseconds.
Fluoride triggered cytotoxicity was studied with standard MTT assays (Fig. 5). Initially, we had to investigate the toxicity of fluoride ions, and find a safe range for the cells. It was determined that fluoride in the form of sodium fluoride, was non-toxic up to 5 mM concentration in cell cultures. Also, the endoperoxide alone had no toxic cytotoxic effects. At 2.5 μM concentration of the endoperoxide 1, the MCF-7 cell cultures were not affected. However, when 5 mM fluoride was added to the cell culture, 65% of the cancer cells were killed. At 5.0 μM endoperoxide and 5 mM fluoride, 85% of the cancer cells were dead. We then wanted to further investigate the cell death with microscopy. Using ROS sensor 2′,7′-dichlorodihydrofluorescein diacetate (DCDHF-DA) to characterize the intracellular release of singlet oxygen, we were able to demonstrate that the combination of endoperoxide and fluoride releases ROS, and considering the fluorescence signal is suppressed in the presence of added azide, the ROS in question is indeed singlet oxygen, as expected (Fig. 6).
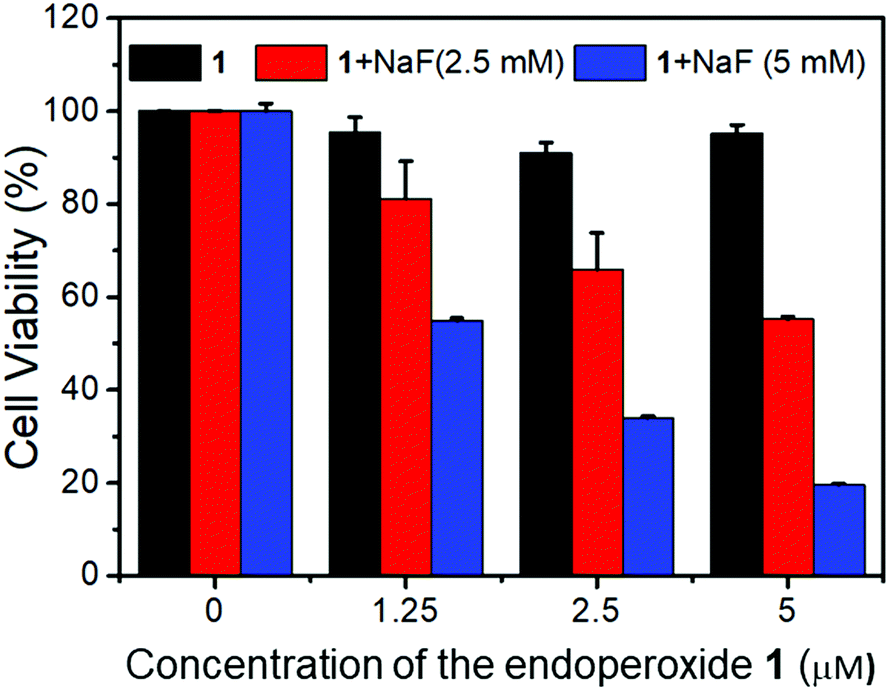 | ||
| Fig. 5 Cell viability as a function of endoperoxide (1) concentration in cell cultures as determined by MTT assays. Black bars indicate cell viability with endoperoxide alone at the indicated concentration, red bars with 2.5 mM fluoride and blue bars 5.0 mM fluoride, with indicated concentrations of the endoperoxide. | ||
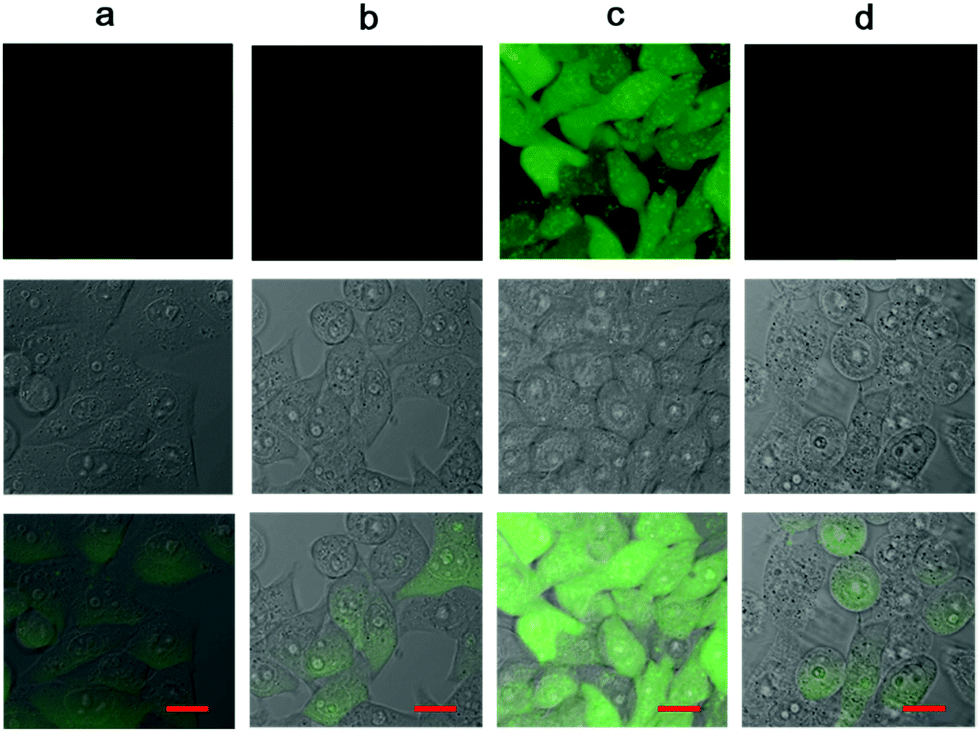 | ||
| Fig. 6 Imaging of the singlet oxygen release with ROS probe DCDHF-DA inside cultured MCF-7 cells. (a) Control cells, (b) incubation with 5 μM endoperoxide 1, (c) incubation with 5 μM endoperoxide 1, and 5 mM NaF, (d) incubation with 5 μM endoperoxide 1, and 5 mM NaF and 10 mM NaN3. Top row fluorescence image, middle row DIC, bottom, merged. Scale bar is 20 μm. | ||
Then, using dual staining with fluorescein-labeled Annexin-V (green emission) and nuclear stain propidium iodide (PI, red emission) we looked for signs of apoptotic cell death (Fig. 7). The confocal microscopy images show a very clear green halo view of the apoptotic membranes labelled with Annexin-V-FITC and concurrently, PI efficiently labels their nuclei demonstrating compromised membrane integrity, only in the presence of 1 and fluoride. This, together with the signs of blebbing and nuclear condensation and fragmentation, clearly demonstrate that MCF-7 cells when subjected to these conditions, undergo apoptosis. Controls show no change, and sodium azide supresses the changes, indicating once again, that the observed differences are due to singlet oxygen. The results also clearly demonstrate that the other product of the cycloreversion reaction, which is 1,4-dimethylnaphthalene, is not toxic at the concentrations used in this work.
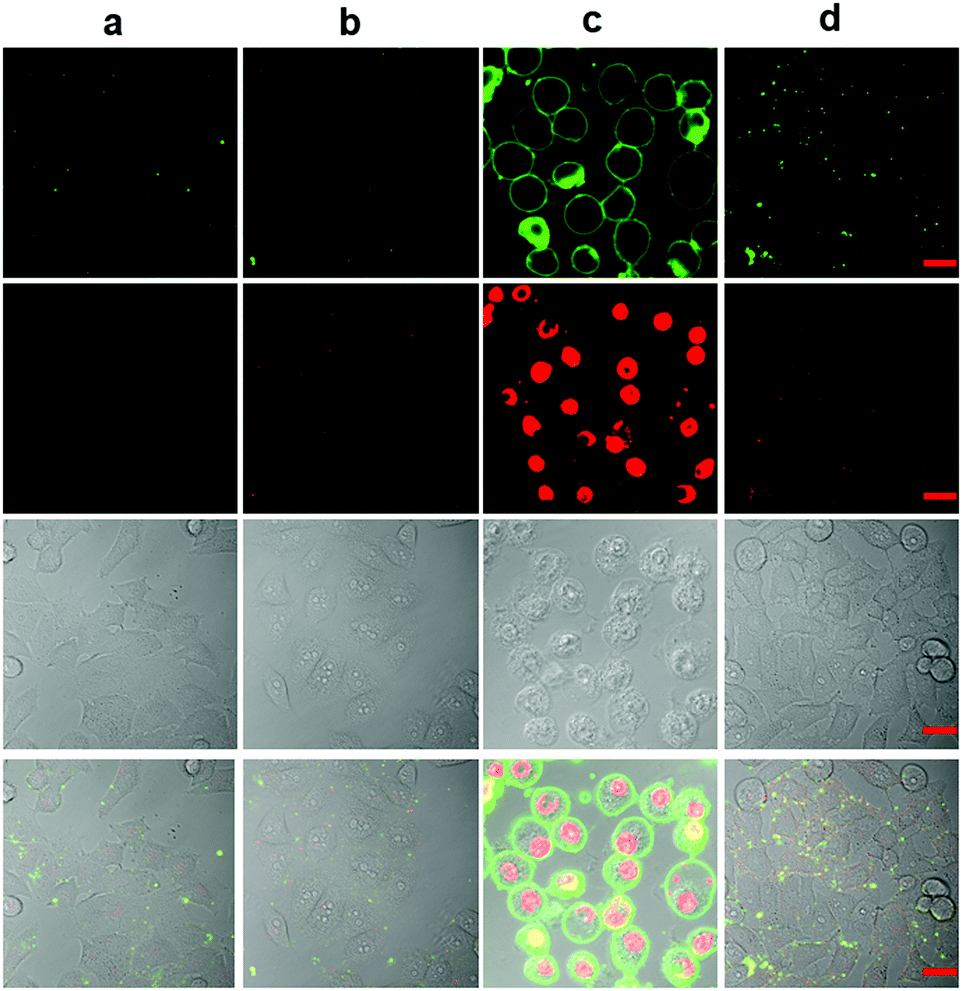 | ||
| Fig. 7 Co-staining with fluorescein conjugate of Annexin V (Annexin V-FITC) and propidium iodide (PI) of MCF-7 cells. (a) Control cells, (b) incubation with 5 μM endoperoxide 1, (c) incubation with 5 μM endoperoxide 1, and 5 mM NaF, (d) incubation with 5 μM endoperoxide 1, and 5 mM NaF and 10 mM NaN3. Top row, fluorescence image (green channel), 2nd (from the top) row fluorescence image (red channel), 3rd (from the top) row DIC, bottom row, merged. Scale bar is 20 μm. | ||
In summary, we present a proof of principle for a two-stage photodynamic action. We demonstrated that it is possible to transform aromatic endoperoxides from a relatively stable from to a labile form in aqueous solutions. Released singlet oxygen from a micromolar concentration of naphthalene endoperoxide is sufficient to induce apoptotic cell death in MCF-7 cell cultures. While fluoride itself is not an ideal stimulus to trigger singlet oxygen release in vivo, the work presented here clearly demonstrates the path towards a new therapeutic paradigm, where the active agent of photodynamic therapy of cancer is generated and delivered, without any need for external oxygen, or light. Our work along that path is in progress.
The authors acknowledge support from Dalian University of Technology, Grant No. DUT18RC(3)062 (E. U. A.) and the National Science Foundation of China, Grant No. 21808028 (W. S.).
Conflicts of interest
There are no conflicts to declare.Notes and references
- X. S. Li, S. Kolemen, J. Yoon and E. U. Akkaya, Adv. Funct. Mater., 2017, 27, 1604053 CrossRef.
- X. S. Li, S. Lee and J. Yoon, Chem. Soc. Rev., 2018, 47, 1174–1188 RSC.
- X. S. Li, N. Kwon, T. Guo, Z. Liu and J. Yoon, Angew. Chem., Int. Ed., 2018, 57, 11522–11531 CrossRef CAS PubMed.
- M. Li, J. Xia, R. Tian, J. Wang, J. Fan, J. Du, S. Long, X. Song, J. W. Foley and X. J. Peng, J. Am. Chem. Soc., 2018, 140, 14851–14859 CrossRef CAS PubMed.
- I. S. Turan, G. Gunaydin, S. Ayan and E. U. Akkaya, Nat. Commun., 2018, 9, 805 CrossRef PubMed.
- S. Callaghan and M. O. Senge, Photochem. Photobiol. Sci., 2018, 17, 1490–1514 RSC.
- Z. Yuan, S. Yu, F. Cao, Z. Mao, C. Gao and J. Ling, Polym. Chem., 2018, 9, 2124–2133 RSC.
- H. Key, E. R. Davies, P. C. Jackson and P. N. T. Wells, Phys. Med. Biol., 1991, 36, 579–590 CrossRef CAS PubMed.
- J. L. Sandell and T. C. Zhu, J. Biophotonics, 2011, 4, 773–787 CrossRef.
- B. Muz, P. de la Puente, F. Azab and A. K. Azab, Hypoxia, 2015, 3, 83–92 CrossRef PubMed.
- P. Vaupel and A. Mayer, Cancer Metastasis Rev., 2007, 26, 225–239 CrossRef CAS PubMed.
- N. Basset-Seguin, Ann. Dermatol. Vener., 2013, 140, 223–228 CrossRef.
- X. Wen, Y. Lin and M. R. Hamblin, Photodiagn. Photodyn. Ther., 2017, 19, 140–152 CrossRef CAS PubMed.
- H. von Tappeiner and A. Jodlebauer, Dtsch. Arch. Klin. Med., 1904, 80, 427–487 Search PubMed.
- S. Kolemen, T. Ozdemir, D. Lee, G. M. Kim, T. Karatas, J. Yoon and E. U. Akkaya, Angew. Chem., Int. Ed., 2016, 55, 3606–3610 CrossRef CAS PubMed.
- I. S. Turan, D. Yildiz, A. Turksoy, G. Gunaydin and E. U. Akkaya, Angew. Chem., Int. Ed., 2016, 55, 2875–2878 CrossRef CAS PubMed.
- T. Ozdemir, Y.-C. Lu, S. Kolemen, E. Tanriverdi-Ecik and E. U. Akkaya, ChemPhotoChem, 2017, 1, 183–187 CrossRef CAS.
- J.-M. Aubry, C. Pierlot, J. Rigaudy and R. Schmidt, Acc. Chem. Res., 2003, 36, 668–675 CrossRef CAS PubMed.
- W. Fudickar and T. Linker, Angew. Chem., Int. Ed., 2018, 57, 12971–12975 CrossRef CAS PubMed.
- H. H. Wasserman, K. B. Wiberg, D. L. Larsen and J. Parr, J. Org. Chem., 2005, 70, 105–109 CrossRef CAS PubMed.
- M. Klaper and T. Linker, Chem. – Eur. J., 2015, 21, 8569–8577 CrossRef CAS PubMed.
- Y. Ito, Y. Amino, M. Nakatsuka and T. Saegusa, J. Am. Chem. Soc., 1983, 105, 1586–1590 CrossRef CAS.
- I. S. Turan and E. U. Akkaya, Org. Lett., 2014, 16, 1680–1683 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9cc06231a |
| This journal is © The Royal Society of Chemistry 2019 |