
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA hydroxylamine probe for profiling S-acylated fatty acids on proteins†
Janine
Schulte-Zweckel
a,
Mridula
Dwivedi
b,
Andreas
Brockmeyer
a,
Petra
Janning
a,
Roland
Winter
b and
Gemma
Triola
 *c
*c
aDepartment of Chemical Biology, Max-Planck-Institute of molecular Physiology, Otto-Hahn-Strasse 11, D-44227 Dortmund, Germany
bFaculty of Chemistry and Chemical Biology, Department of Physical Chemistry, Technical University of Dortmund, Otto-Hahn-Str.4a, 44221 Dortmund, Germany
cDepartment of Biological Chemistry, Institute of Advanced Chemistry of Catalonia (IQAC-CSIC), Jordi Girona 18–26, 08034 Barcelona, Spain. E-mail: gemma.triola@iqac.csic.es
First published on 29th August 2019
Abstract
Reversible S-palmitoylation is a key regulatory mechanism of protein function and localization. There is increasing evidence that S-acylation is not restricted to palmitate but it includes shorter, longer, and unsaturated fatty acids. However, the diversity of this protein modification has not been fully explored. Herein, we report a chemical probe that combined with MS-based analysis allows the rapid detection and quantification of fatty acids linked to proteins. We have used this approach to profile the S-acylome and to show that the oncogene N-Ras is heterogeneously acylated with palmitate and palmitoleate. Studies on protein distribution in membrane subdomains with semisynthetic proteins revealed that unsaturated N-Ras presents an increased tendency toward clustering and higher insertion kinetic rate constants.
The acylation of cysteinyl residues with fatty acids, also termed S-acylation, is one of the most common lipid post-translational modifications (PTM) of proteins. This reversible modification can occur on soluble and transmembrane proteins and has an important role in the regulation of various cellular functions, including protein localization, stability, trafficking, and interaction with effectors.1
S-Acylation is also known as S-palmitoylation because the 16-carbon saturated palmitic acid is the most commonly attached lipid. However, there is now accumulating evidence that fatty acids of various chain lengths and degrees of unsaturation can modify lipoproteins thereby suggesting that heterogeneous S-acylation may have an additional role in the regulation of protein localization and function. Hence, the pool of circulating free fatty acids comprise more than 40 distinct molecular species.2 In addition, Wnt protein has been found to be O-acylated with palmitoleic acid,3 and stearic or arachidonic acid have been occasionally detected in S-acylated proteins.4–6 Most of these previous studies have been performed by metabolic labeling with azide/alkyne-tagged7 or radioactive fatty acids.4
However, these exogenously added lipids can be metabolized to shorter, longer or unsaturated fatty acids thereby precluding the confident identification of lipid residues in the native state. Thioesters can be selectively cleaved by treatment with hydroxylamine resulting in the generation of a free sulfhydryl and the conversion of the released fatty acid into fatty acid hydroxamates (FAH) (Fig. 1A). This selective hydrolysis has already been exploited in the acyl-biotin exchange technique to identify lipidated proteins. However, examples of using this approach to identify the released fatty acids are scarce. The standard method is based on gas chromatography-mass spectrometry (GC-MS), requires the previous conversion of hydroxamates to esters and can only be used to assess gross changes in protein acylation.5,8 This technique has also revealed that Rho related GTPase (ROP6) and the calcium sensor protein CBL1 were both modified with palmitic and stearic acid. However, this approach requires at least 1 μg of purified protein, which makes it not practicable as a high-throughput method.9 Consequently, this field of research could strongly benefit from novel chemical tools and robust and general methods enabling the direct and sensitive analysis of S-linked fatty acids. Nowadays, GC-MS analysis of lipids has been mainly overtaken by high-resolution MS coupled to liquid chromatography (LC), showing the ability to resolve, profile and accurately identify multiple lipid species. As a result, our main aim was to develop an appropriate LC-HRMS method for accurate FAH analysis.
 | ||
| Fig. 1 (A) General workflow and structure of the hydroxylamine probes and the resulting fatty acid hydroxamates. (B) Quantification of the S-acyl chains attached to proteins was performed in HEK293, HeLa and N2a cells after treatment with hydroxylamine probe 2. (C) HEK293 and N2a cells after incubation with a SCD1 inhibitor and treatment with hydroxylamine derivative 2. Values shown are mean ± SD (n = 5). | ||
We initially synthesized the palmitic acid hydroxamate 1 and investigated its performance. FAH need to be soluble in organic solvents to ensure its proper extraction from lysate samples and subsequent analysis. However, the known low solubility of 1 in organic solvents10,11 complicated its handling, isolation from lysates and its LC-HRMS-based analysis. Since functionalized hydroxylamines have shown to retain the ability to cleave thioesters,12 we designed the probe 2 bearing a fluorescein moiety. We also synthesized the resulting palmitic acid hydroxamate 3 that showed a higher solubility in organic solvents and a better performance in LC-HRMS-based analysis compared to 1. Moreover, the presence of the fluorescein may facilitate its use in future analytical methods based on HPLC and fluorescence detection. Next, several additional FAHs derived from 2 (12:0, 14:0, 16:1, 18:0, 18:1, 20:0, 20:4) were synthesized as reference compounds. Linearity and robustness of their MS detection was assessed (limit of quantification of ∼100 fmol and linear response up to 25 pmol, see ESI†). MS analysis after incubation of a palmitoylated cysteine with 2 confirmed dose-dependent formation of the corresponding FAH.
We then sought to establish whether 2 can be used for the identification of endogenous S-acyl chains. To this end, the proteome of HEK293, HeLa and Neuro 2A (N2a) cells was precipitated, and lipids non-covalently associated with proteins were removed by washing the protein pellet with chloroform/methanol. The proteome was then treated with 2 and the released fatty acids hydroxamates were extracted and analyzed by LC-HRMS. The non-natural heptadecanoic acid (C17:0) was spiked into the samples at a fixed concentration for relative quantification. The identity of the formed fatty acid hydroxamates was unambiguously confirmed by accurate mass and elemental composition and by comparing their retention time with the ones observed for synthesized reference compounds. These studies revealed an important diversity and substantial cell type-specific differences (Fig. 1B). Hence, in HEK293 and HeLa cells, proteins S-acylated with palmitic (C16:0), palmitoleic (16:1) and oleic (18:1) acids were the most abundant ones. In contrast, N2a cells contain only traces of palmitoleic acid whereas the amount of stearic (18:0) acid detected in these cells was markedly higher than in the other cell lines. In all the cases only low levels of myristic (14:0), linoleic (18:2) and arachidic acid (20:0) could be observed, while no signal of modification with lauric (12:0) and arachidonic (20:4) acid could be detected. FAH could only be detected in cells treated with 2. We next investigated if an exogenously added fatty acid could modify the S-acylome. Thus, cells were fed with the non-natural fatty acid nonadecanoic acid (C19:0), and after MS-analysis of thioester linked lipids, the corresponding FAH could be detected in all the cell lines tested (see Table S1, ESI†), accompanied by a decrease in the amount of all the other fatty acids.
Unsaturated fatty acids are synthesized from saturated precursors by means of fatty acid desaturases such as stearoyl-Coenzyme A desaturase 1 (SCD1), which catalyzes the conversion of stearate (18:0) to oleate (18:1) and of palmitate (16:0) to palmitoleate (16:1). To explore the role of SCD1 in the synthesis of proteins modified with unsaturated fatty acids, HEK293 and N2a cells were treated with a SCD1 inhibitor prior to analysis by MS.13 Indeed, there was a significant decrease in the desaturation index of treated cells, defined as the ratio of C16:1 to C16:0 or C18:1 to C18:0 (Fig. 1C). However, a substantial amount of unsaturated fatty acids remained yet bound to the proteins under these conditions, thereby suggesting that alternative mechanisms might coexist to convert saturated to unsaturated fatty acids prior to or after transfer to proteins.
Encouraged by the high sensitivity, we then extended our studies to the analysis of the S-fatty acids attached to the small GTPase N-Ras, which undergo a reversible posttranslational S-palmitoylation that govern its subcellular localization and function.14 With this aim, HEK293 cells and N2a cells were stably transfected with a vector carrying HA-N-Ras or eGFP-N-Ras, respectively. Transfection efficiency was assessed by measuring the gene expression through real-time quantitative PCR (qPCR, Fig. 2A) and Western blot analysis was employed to determine the relative expression of the transfected protein using α-Tubulin as an internal control (Fig. 2B). Moreover, direct identification of N2a transfected cells could be achieved upon microscopy visualization of eGFP-N-Ras (Fig. 2C). Overexpressed N-Ras protein was then isolated by antibody immune affinity purification using either anti-HA or anti-eGFP-modified agarose. Protein fraction bound to agarose was incubated with the hydroxylamine probe 2 and the released fatty acid hydroxamates were analysed by HRMS as described before. Results indicate that both cell lines contained N-Ras modified with both palmitic acid (C16:0, 65%) and the unsaturated palmitoleic acid (C16:1; 35%) (Fig. 3D), whereas no traces of other fatty acids could be detected. In agreement with these results, N-Ras was recently shown to get acylated when treating cells with a ω-alkynyl palmitoleic acid.7
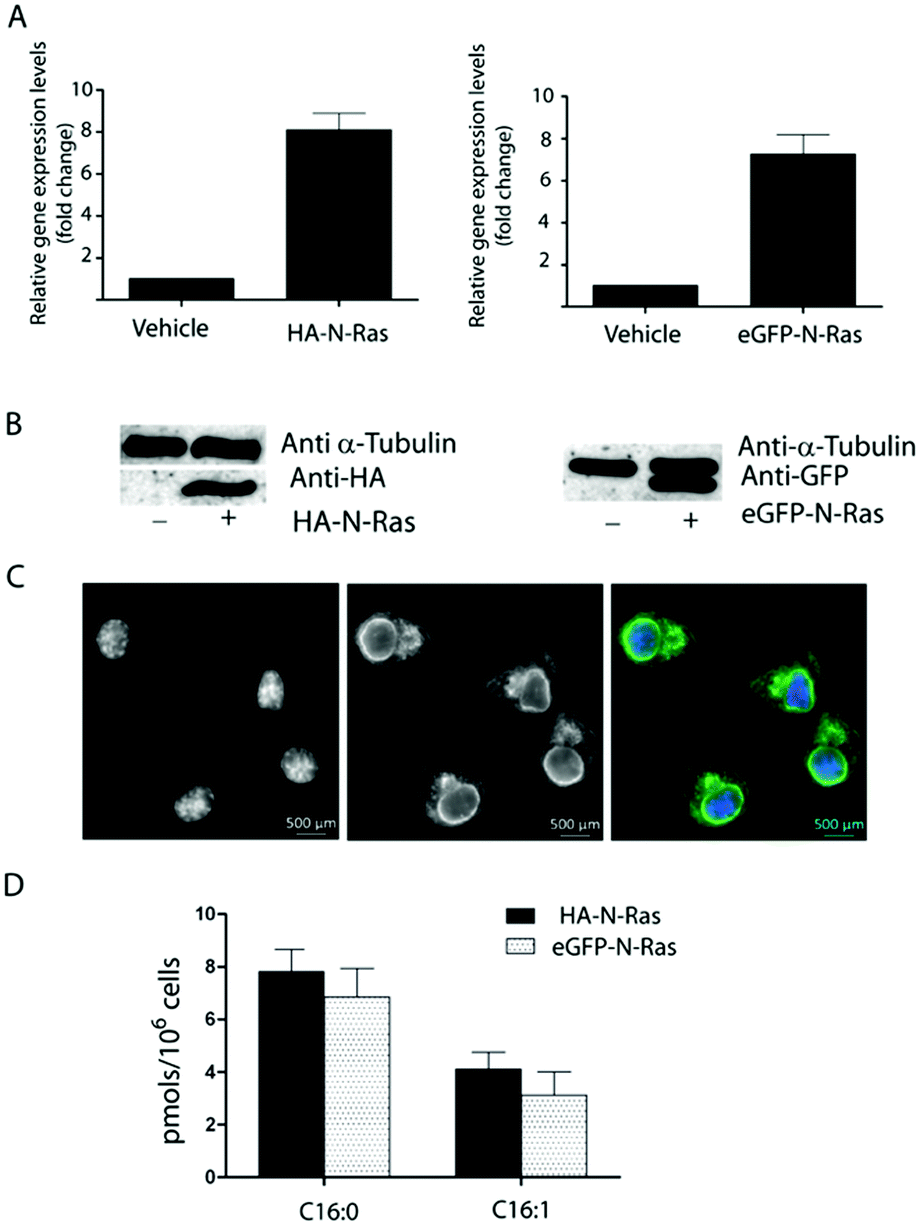 | ||
| Fig. 2 (A) Relative expression levels measured by qPCR. (B) Western blot analysis of the overexpression of HA-N-Ras and eGFP-N-Ras. Left: α-Tubulin and anti-HA antibody; right: α-Tubulin and anti-eGFP antibody. (C) Stable transfection of eGFP-N-Ras in N2a cells. Left: DAPI-stained nucleus; middle: eGFP-N-Ras; right: Merged channels. (D) Quantification of the S-acyl chains attached to N-Ras was performed in HEK293 cells overexpressing HA-N-Ras and N2a cells overexpressing eGFP-N-Ras after treatment with fluorescein–ONH23. Values shown are mean ± SD (n = 5) | ||
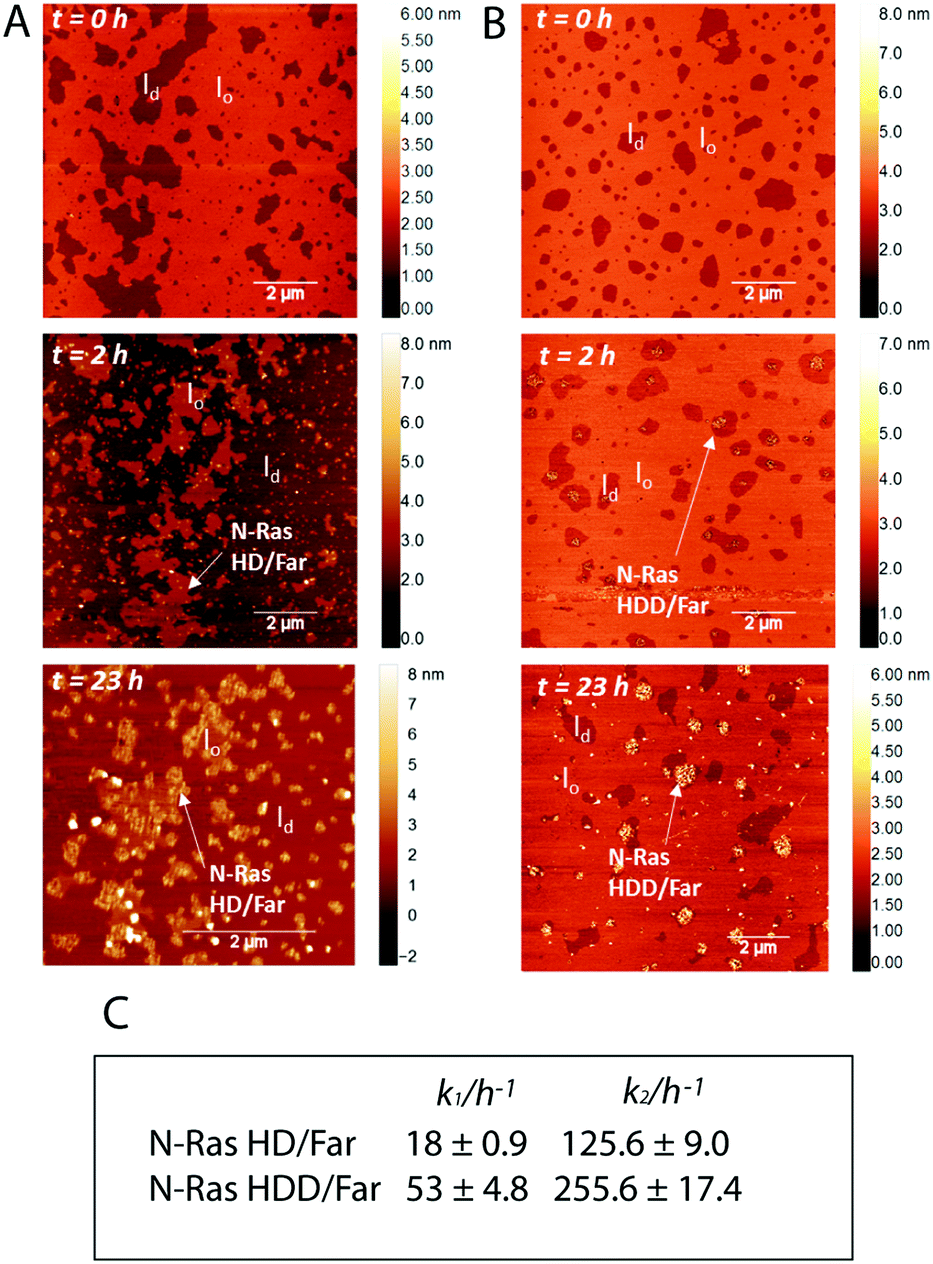 | ||
Fig. 3 Time-lapse AFM scans upon insertion of 4.5 μM (A) N-Ras HD/far and (B) N-Ras HDD/far isoforms into lipid bilayers composed of the zwitterionic ‘raft’ membrane DOPC/DPPC/chol (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:1) which forms coexisting liquid-ordered (lo) and liquid-disordered (ld) domains (ratio protein:lipid = 1:118). (C) Kinetic rate constants for zwitterionic “raft” membrane association of N-Ras HD/far and N-Ras HDD/far at 25 °C. :2:1) which forms coexisting liquid-ordered (lo) and liquid-disordered (ld) domains (ratio protein:lipid = 1:118). (C) Kinetic rate constants for zwitterionic “raft” membrane association of N-Ras HD/far and N-Ras HDD/far at 25 °C. | ||
Heterogenous S-acylation may have a crucial impact on the reversible membrane binding of soluble proteins, and can influence membrane partitioning, trafficking and signalling processes. Thus, acylation of the HIV protein Gag with unsaturated fatty acids results in loss of raft association and inability to mediate viral particle assembly.15 Similarly, the different lipidation pattern of K-, H- and N-Ras proteins determine distinct intracellular localization (Golgi vs. plasma membrane)16 and different partitioning in membrane microdomains.17–19 Moreover, palmitoleic acid is a tightly regulated signalling lipid that controls several metabolic activities20 and the unsaturated double bond, which creates a kink in the fatty acyl chain, may influence protein interaction with cellular membranes. To explore if the two populations of lipidated N-Ras exhibit a different behaviour, we prepared two semisynthetic farnesylated N-Ras proteins bearing a palmitoyl or a palmitoleoyl anchors by means of maleimide ligation of a truncated N-Ras to a lipidated peptide (see ESI†).17 Using this approach, we could obtain two doubly lipidated N-Ras proteins bearing a farnesyl group and nonhydrolizable mimics of the palmitoyl (N-Ras HD/far) or the palmitoleoyl anchors (N-Ras HDD/far). To investigate the effect of saturation of the lipid acyl chain dependent partitioning of N-Ras protein, we performed high resolution membrane partitioning studies using atomic force microscopy. A heterogeneous zwitterionic solid supported bilayer was prepared with the composition of DOPC/DPPC/chol (1:2:1). Under ambient conditions, this composition forms co-existence of liquid-ordered (lo) and liquid-disordered (ld) domains, reminiscent of membrane “rafts”. The coexistence of the domains was confirmed at the beginning of the experiment using time-lapse tapping mode in fluid AFM. After addition of the N-Ras protein, the bilayer was incubated for 2 h, and the unbound protein was then subsequently washed away with buffer solution. As shown in Fig. 3A, N-Ras HD/far protein localizes and completely fills the ld phase. After 23 h, it localizes and clusters largely at the ld/lo phase boundaries, in agreement with our previous studies.17 On the other hand, N-Ras HDD/far was shown to localize and cluster in the liquid-disordered domains, and this scenario did not change significantly over 23 h (Fig. 3B), indicating a different partitioning behaviour compared to the saturated N-Ras HD/far isoform. Previously, it was shown that the lipid anchors of the N-Ras proteins are capable of adjusting their length to match its surrounding lipid matrix.21 This in turn increases the packing efficiency of the inserted acyl chain and prevents packing defects. The presence of an unsaturation in the acyl chain may decrease the ability of the acyl chain for efficient packing thus introducing packing defects. As a consequence, owing to a membrane-mediated effect, the unsaturated N-Ras HDD/far forms large stable clusters in the fluid lipid phase. As has been previously shown, the insertion and clustering of the lipidated signaling proteins in membranes is actively driven by the lipid anchors of the proteins either through electrostatic interactions22,23 or through the hydrophobic effect (hydrophobic mismatch).17 Our results reveal that unsaturation in the acyl chain can also influence the clustering behavior of lipidated Ras proteins owing to the decreased packing efficiency of the unsaturated acyl chain in the lipid matrix. As a consequence of the perturbation of lipid packing, the tendency toward clustering increases, which reduces the overall free energy of the proteolipid system.
To reveal also the effect of saturation of the acyl chain on the insertion kinetics of the lipidated proteins, we performed a fluorescence resonance energy transfer (FRET) based kinetics assay. Large unilamellar vesicles (LUVs) composed of DOPC/DPPC/chol (1:2:1) were labelled with 0.1 mol% rhodamine DHPE. Insertion and binding of Bodipy labelled N-Ras protein led to an increase in the FRET intensity as a function of time. The kinetic data were found to be best fitted using a biexponential association kinetics, i.e. insertion of the N-Ras protein proceeds via a two-step process (the corresponding rate constants are summarized in Fig. 3C). This is in agreement with previous studies which proposed the two steps to be the insertion of the acyl chain into the membrane which is followed by lateral rearrangement of the protein in the membrane plane to attain the most stable equilibrium state.24 Interestingly, the kinetic rate constants of both the steps were found to be significantly higher for the unsaturated N-Ras HDD/far than for the saturated N-Ras HD/far protein, indicating a decreased activation energy for the insertion and the lateral rearrangement process. This may be attributed to the different oligomerization states of the N-Ras isoforms in the bulk which may further influence the membrane insertion energies. This is an interesting result because this indicates that the kinetics of the membrane partitioning process of lipidated proteins can be regulated by changing the saturation state of the acyl chains, along with other molecular parameters such as the charge density of the hypervariable region of the Ras protein.
To conclude, we have developed a chemical probe that enables the accurate and systematic identification of S-bound fatty acids in their native state. A spiked internal standard allows the relative quantification of the fatty acids. The analysis of the S-acylome in different cell lines as well as in an enriched N-Ras revealed that proteins are modified with fatty acids of various chain lengths and structures. These previously overlooked heterogenous acyl chains may have an additional role in regulating protein localization and function. Thus, AFM studies and FRET-based kinetics assays indicated that the unsaturated N-Ras presents an increased tendency toward clustering and higher insertion kinetic rate constants compared to that of its saturated counterpart. Unfortunately, our current method as well as direct LC-MS/MS analysis25 or metabolic labelling,4,7,26 still require overexpression and purification of individual proteins. Alternative hydroxylamine derivatives will be explored in the future to overcome this limitation. All in all, we are convinced that the combined use of hydroxylamine derivatives and HRMS-based analysis may strongly contribute to a better understanding of the biochemistry of S-acylated proteins.
This work was supported by the Max Planck Society (Max Planck Partner Group to G. T.) with partial support from the Ministerio de Economia and Competitividad (grant CTQ2013-44334-P). We acknowledge support of the publication fee by the CSIC Open Access Publication Support Initiative through its Unit of Information Resources for Research (URICI).
Conflicts of interest
There are no conflicts to declare.Notes and references
- L. H. Chamberlain and M. J. Shipston, Physiol. Rev., 2015, 95, 341 CrossRef CAS PubMed.
- O. Quehenberger, A. M. Armando, A. H. Brown, S. B. Milne, D. S. Myers, A. H. Merrill, S. Bandyopadhyay, K. N. Jones, S. Kelly, R. L. Shaner, C. M. Sullards, E. Wang, R. C. Murphy, R. M. Barkley, T. J. Leiker, C. R. Raetz, Z. Guan, G. M. Laird, D. A. Six, D. W. Russell, J. G. McDonald, S. Subramaniam, E. Fahy and E. A. Dennis, J. Lipid Res., 2010, 51, 3299 CrossRef CAS PubMed.
- R. Takada, Y. Satomi, T. Kurata, N. Ueno, S. Norioka, H. Kondoh, T. Takao and S. Takada, Dev. Cell, 2006, 11, 791 CrossRef CAS PubMed.
- X. Q. Liang, Y. Lu, T. A. Neubert and M. D. Resh, J. Biol. Chem., 2002, 277, 33032 CrossRef CAS PubMed.
- L. Muszbek, G. Haramura, J. E. Cluette-Brown, E. M. Van Cott and M. Laposata, Lipids, 1999, 34, S331 CrossRef CAS PubMed.
- K. Brett, L. V. Kordyukova, M. V. Serebryakova, R. R. Mintaev, A. V. Alexeevski and M. Veit, J. Biol. Chem., 2014, 289, 34978 CrossRef PubMed.
- B. Zheng, G. K. Jarugumilli, B. Chen and X. Wu, ChemBioChem, 2016, 17, 2022 CrossRef CAS PubMed.
- F. Mohammadzadeh, V. Hosseini, A. Mehdizadeh, C. Dani and M. Darabi, IUBMB Life, 2018, 71, 340 CrossRef PubMed.
- N. Sorek, A. Akerman and S. Yalovsky, Methods Mol. Biol., 2013, 1043, 121 CrossRef CAS PubMed.
- I. S. Rosenfeld, G. D'Agnolo and P. R. Vagelos, Anal. Biochem., 1975, 64, 221 CrossRef CAS PubMed.
- F. Vernon and J. H. Khorassani, Talanta, 1978, 25, 410 CrossRef CAS PubMed.
- C. Sarkar, G. Chandra, S. Peng, Z. Zhang, A. Liu and A. B. Mukherjee, Nat. Neurosci., 2013, 16, 1608 CrossRef CAS PubMed.
- G. Liu, J. K. Lynch, J. Freeman, B. Liu, Z. Xin, H. Zhao, M. D. Serby, P. R. Kym, T. S. Suhar, H. T. Smith, N. Cao, R. Yang, R. S. Janis, J. A. Krauser, S. P. Cepa, D. W. Beno, H. L. Sham, C. A. Collins, T. K. Surowy and H. S. Camp, J. Med. Chem., 2007, 50, 3086 CrossRef CAS PubMed.
- O. Rocks, A. Peyker, M. Kahms, P. J. Verveer, C. Koerner, M. Lumbierres, J. Kuhlmann, H. Waldmann, A. Wittinghofer and P. I. H. Bastiaens, Science, 2005, 307, 1746 CrossRef CAS PubMed.
- O. W. Lindwasser and M. D. Resh, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 13037 CrossRef CAS PubMed.
- O. Rocks, M. Gerauer, N. Vartak, S. Koch, Z. P. Huang, M. Pechlivanis, J. Kuhlmann, L. Brunsveld, A. Chandra, B. Ellinger, H. Waldmann and P. I. H. Bastiaens, Cell, 2010, 141, 458 CrossRef CAS PubMed.
- K. Weise, G. Triola, L. Brunsveld, H. Waldmann and R. Winter, J. Am. Chem. Soc., 2009, 131, 1557 CrossRef CAS PubMed.
- Y. X. Chen, S. Koch, K. Uhlenbrock, K. Weise, D. Das, L. Gremer, L. Brunsveld, A. Wittinghofer, R. Winter, G. Triola and H. Waldmann, Angew. Chem., Int. Ed., 2010, 49, 6090 CrossRef CAS PubMed.
- K. Weise, S. Kapoor, C. Denter, J. Nikolaus, N. Opitz, S. Koch, G. Triola, A. Herrmann, H. Waldmann and R. Winter, J. Am. Chem. Soc., 2011, 133, 880 CrossRef CAS PubMed.
- H. Cao, K. Gerhold, J. R. Mayers, M. M. Wiest, S. M. Watkins and G. S. Hotamisligil, Cell, 2008, 134, 933 CrossRef CAS PubMed.
- A. Vogel, G. Reuther, K. Weise, G. Triola, J. Nikolaus, K. T. Tan, C. Nowak, A. Herrmann, H. Waldmann, R. Winter and D. Huster, Angew. Chem., Int. Ed., 2009, 48, 8784 CrossRef CAS PubMed.
- C. Nicolini, J. Baranski, S. Schlummer, J. Palomo, M. Lumbierres-Burgues, M. Kahms, J. Kuhlmann, S. Sanchez, E. Gratton, H. Waldmann and R. Winter, J. Am. Chem. Soc., 2006, 128, 192 CrossRef CAS PubMed.
- M. Dwivedi, T. Mejuch, H. Waldmann and R. Winter, Angew. Chem., Int. Ed., 2017, 56, 10511 CrossRef CAS PubMed.
- S. Kapoor, A. Werkmuller, R. S. Goody, H. Waldmann and R. Winter, J. Am. Chem. Soc., 2013, 135, 6149 CrossRef CAS PubMed.
- E. Thinon, J. P. Fernandez, H. Molina and H. C. Hang, J. Proteome Res., 2018, 17, 1907 CrossRef CAS PubMed.
- E. Thinon, A. Percher and H. C. Hang, ChemBioChem, 2016, 17, 1800 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, figures and tables. See DOI: 10.1039/c9cc05989j |
| This journal is © The Royal Society of Chemistry 2019 |