
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelf-templated synthesis of an orthoformate in,in-cryptand and its bridgehead inversion by dynamic covalent exchange†
Henrik
Löw
,
Elena
Mena-Osteritz
and
Max
von Delius
 *
*
Institute of Organic Chemistry, University of Ulm, Albert-Einstein-Allee 11, 89081 Ulm, Germany. E-mail: max.vondelius@uni-ulm.de
First published on 4th September 2019
Abstract
We report the template-free dynamic covalent self-assembly of a small orthoformate cryptand, which appears to be driven by the formation of two sets of intramolecular, four-centre hydrogen bonds. In contrast to their nitrogen-bridgehead counterparts, orthoformate cryptands do not spontaneously invert, but require dynamic covalent exchange to do so.
The in,out-isomerism of macrocycles and cryptands has been examined in much detail during the second half of the last century.1 Early examples2 included katapinands,3 protonated azacryptands4 as well as phosphor-bridged5 macrocycles and cryptands. Mechanistically, bridgehead inversion was found to occur by homeomorphic isomerisation,1,2a which inverts the configuration at the bridgehead atoms by pulling one chain through the macrocycle defined by the other two chains.6 This process was found to either require protonation/deprotonation of the Brønsted basic bridgehead atoms or large ring sizes (n > 20).1,2a To the best of our knowledge, there are no reports on the inversion of macrobicyclic architectures by dynamic covalent chemistry (DCC).
We have recently made use of the dynamic covalent reaction7 between orthoesters and diols8 to prepare a new class of monometallic cryptands.9 During extensive studies on the scope of these templated self-assembly reactions (Scheme 1a),9a we observed that orthoformates are unique outliers, because they are the only out,out-cryptands templated by the Li+ ion (normally: Na+). Herein, we report a more fundamental facet of this outlier behaviour, namely that in the absence of metal ions, a remarkable in,in-cryptand is formed in a process that appears to be driven by intramolecular hydrogen bonds centred on the positively polarised hydrogen atoms of the orthoformate bridgeheads. With the metal-templated out,out-cryptate and the self-templated in,in-cryptand in hand, we were able to achieve inversion at the carbon bridgeheads by means of acid-catalysed dynamic covalent exchange (Scheme 1b).
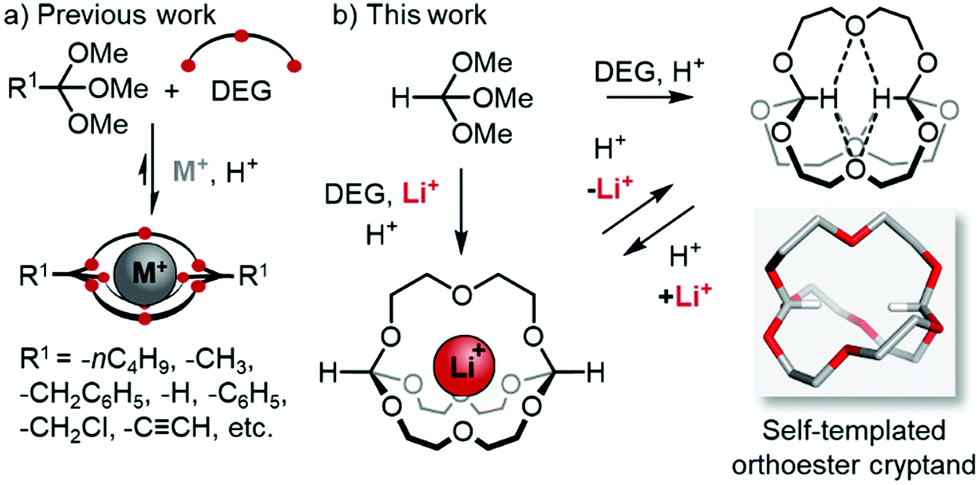 | ||
| Scheme 1 (a) Previous work on the scope of self-assembled orthoester cryptands. (b) Syntheses of two orthoformate cryptands: Li+-templated out,out-cryptand, self-templated in,in-cryptand and their interconversion by dynamic covalent bridgehead inversion. | ||
The starting point for the present study was the unexpected observation that the reaction of trimethyl orthoformate and DEG with 1% TFA as catalyst in the absence of a metal template furnished orthoester cryptand o-(Hin)2-1.1.1 in ca. 50% isolated yield (Scheme 2). This result was surprising, because we had previously found9a (Scheme 1a) that a suitable alkali metal template (e.g. Li or NaBArF) is strictly necessary to avoid the predominant formation of eight-membered ring products.9a,b
 | ||
| Scheme 2 Interconversion of self-templated in,in- and metal-templated out,out-orthoformate cryptands. Percent values indicate isolated yields. Reaction conditions: (i) trimethyl orthoformate, diethylene glycol and TFA in CHCl3, 5 Å molecular sieves (MS), 3 d. (ii) Lithium tetrakis[3,5-bis(trifluoromethyl)phenyl]borate (LiBArF) or NaBArF, trimethyl orthoformate, diethylene glycol and TFA in CHCl3, 5 Å MS, 3 d. (iii) o-(Hin)2-1.1.1, LiBArF and TFA in CHCl3, 10 min. (iv) o-(Hin)2-1.1.1, NaBArF and TFA in CHCl3, 3 d. (v) o-(H)2-1.1.1 and TFA in CHCl3, 10 min. (vi) [Li+⊂o-(H)2-1.1.1] or [Na+⊂o-(H)2-1.1.1] and chloride-loaded anion exchange resin (Lewatit MP-68) in CHCl3, 12 h. (vii) o-(H)2-1.1.1 and LiBArF or NaBArF in CH3CN, 5 min. For further details, see ESI.† | ||
Conveniently, we were able to obtain highly pure, crystalline material of o-(Hin)2-1.1.1 by simple washing with diethyl ether (see ESI†). It occurred to us that the relatively clean formation of this particular cryptand from a dynamic combinatorial network comprising a multitude of other products may be a result of the formation of intramolecular hydrogen bonds between the bridgehead hydrogen atoms and the central oxygen atoms in the DEG chains. According to this hypothesis, o-(Hin)2-1.1.1 would not be formed by a metal-template effect but rather be “self-templated” (vide infra for X-ray crystallography).
To test this hypothesis, we attempted the transformation of out,out-cryptand o-(H)2-1.1.1‡ into in,in-cryptand o-(Hin)2-1.1.1. A significant difference in free energy between these stereoisomers would suggest that one of the two isomers is subject to thermodynamic stabilisation. While the thermal inversion did not occur under the tested conditions (150 °C, DMF; 200 °C, neat), we found that bridgehead inversion was indeed possible and highly efficient under constitutionally dynamic conditions. As shown in Scheme 2 (bottom), addition of catalytic amounts of acid to o-(H)2-1.1.1 resulted in the quantitative formation of o-(Hin)2-1.1.1 within 5 min.
To demonstrate that the inversion from the out,out- to the in,in-isomer is not due to strain in the out,out-cryptand, we also studied the inversion in the opposite direction. As shown in Scheme 2 (left hand side), this transformation from self-templated o-(Hin)2-1.1.1 to metal-templated [Li+⊂o-(H)2-1.1.1] was also feasible. Only 10 min after 1% acid catalyst and one equivalent LiBArF template was added, the desired out,out-cryptand [Li+⊂o-(H)2-1.1.1] was obtained in 83%. Again, we tested whether a (thermal) homeomorphic isomerisation could be induced by the addition of one equivalent LiBArF as metal template and heating (80 °C, MeCN). However, such an inversion did not occur, demonstrating that one DEG chain cannot be “pulled through” the cryptand to invert the configuration of the bridgeheads.1,2a
Having obtained a preliminary understanding of self-templation and bidirectional bridgehead inversion (left hand side of Scheme 2), we decided to investigate the anomalous9a metal ion affinity of o-(H)2-1.1.1 in more detail (right hand side of Scheme 2). To this end, we initially found that Na+ exchange between o-(H)2-1.1.1 and the bulk is fast on the NMR timescale in chloroform, which is in stark contrast to all other previously investigated orthoester cryptands.9a,c,10 This finding allowed us, for the first time, to study the thermodynamics of sodium binding in the solvent used for the self-assembly reactions. Therefore, we performed triplicate 1H NMR titrations in chloroform and fitted the binding isotherms with a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 stoichiometric model.§ These experiments revealed that the binding of sodium to o-(H)2-1.1.1 in chloroform is rather weak (Ka = 500 M−1 ± 100, see ESI†), indeed about two orders of magnitude weaker than the interaction between Na+ and the methyl-substituted cryptand o-(CH3)2-1.1.110 in the more polar solvent acetonitrile. This explains why we were neither able to self-assemble [Na+⊂o-(H)2-1.1.1] from a mixture of orthoester and diol (Scheme 2, top right) nor obtain the same compound by dynamic covalent bridgehead inversion (Scheme 2, centre right). Nevertheless, we were able to isolate [Na+⊂o-(H)2-1.1.1] by addition of one equivalent of NaBArF to empty o-(H)2-1.1.1 in acetonitrile (Scheme 2, bottom right).
:1 stoichiometric model.§ These experiments revealed that the binding of sodium to o-(H)2-1.1.1 in chloroform is rather weak (Ka = 500 M−1 ± 100, see ESI†), indeed about two orders of magnitude weaker than the interaction between Na+ and the methyl-substituted cryptand o-(CH3)2-1.1.110 in the more polar solvent acetonitrile. This explains why we were neither able to self-assemble [Na+⊂o-(H)2-1.1.1] from a mixture of orthoester and diol (Scheme 2, top right) nor obtain the same compound by dynamic covalent bridgehead inversion (Scheme 2, centre right). Nevertheless, we were able to isolate [Na+⊂o-(H)2-1.1.1] by addition of one equivalent of NaBArF to empty o-(H)2-1.1.1 in acetonitrile (Scheme 2, bottom right).
To obtain structural insights into o-(Hin)2-1.1.1 by X-ray crystallography, we grew single crystals of this compound by slow evaporation of diethyl ether (space group P21/c). The in,in-cryptand has C3h-symmetry and, as expected, does not feature an alkali metal ion at the centre of the cavity (Chart 1a). Instead, both methine hydrogen atoms point directly into the cavity, while the two orthoester carbon atoms have a distance of 4.5 Å. Although X-ray diffraction is not suited to elucidate the precise location of hydrogen atoms, we performed a difference electron density map at a late stage of the structural refinement (OLEX)11 in order to place the methine hydrogen atoms more precisely in the atomistic model. Keeping this limitation in mind, the hydrogen positions are providing a valuable basis for structural insights. Both the approximate H⋯O distances (2.4 to 2.5 Å, sum of van der Waals radii12 = 2.6 Å) and the hydrogen bond angles (C–H⋯O angles: 119 to 122°) point towards the formation of two four-centre hydrogen bonds between the two hydrogen and the three oxygen atoms (Chart 1b). The two bridgehead hydrogen atoms have an approximate distance of 2.5 Å, which is significantly above the sum of van der Waals radii12 (2.2 Å). An analysis of all O–C–O angles of the two orthoester bridgeheads reveals a very narrow distribution (107.5° to 108.2°), which seems unlikely to stem only from packing effects and hence provides support for a rigidification of the molecule by hydrogen bonding.
 | ||
| Chart 1 Solid-state structures of self-templated orthoformate cryptand and two metal-templated orthoformate cryptates. Hydrogen atoms, anions, solvent and disorder (where applicable) are omitted for clarity. Metal ions are displayed at 100% of effective ionic radius.13 (a) Single crystals were obtained by slow evaporation of diethyl ether. Crystal system: monoclinic. (b) Approximate intramolecular hydrogen bond distances and angles in o-(Hin)2-1.1.1 (values received from X-ray crystallography). Oxygen and hydrogen atoms involved in intramolecular hydrogen bonding are highlighted. H⋯O distance: 2.4–2.5 Å. C–H⋯O angles: 119–122°. (c) Single crystals were obtained by the layering method (hexane/chloroform). Crystal system: orthorhombic. (d) The solid-state structure of [Li+⊂o-(H)2-1.1.1] was reported previously9a and is shown for comparison. (e) Comparison of average R–C–O–M torsion angles (coloured dots) of orthoformate and orthoacetate cryptands, including standard deviation and corresponding Newman projections. For further details, see ESI.† | ||
Additionally, we were able to grow single-crystals of the [Na+⊂o-(H)2-1.1.1] cryptate and obtain high-quality X-ray crystallographic data (space group P212121, Chart 1c). While the previously reported complex [Li+⊂o-(H)2-1.1.1] featured only five Li–O bonds (1.96 to 2.23 Å, Chart 1d),9a in [Na+⊂o-(H)2-1.1.1] eight Na–O contacts are observed (2.24 to 2.68 Å), and one orthoester oxygen does not bind the sodium ion (3.61 Å; highlighted in Chart 1c). Although this could in principle be a packing effect, we believe this stems from the previously mentioned outlier behaviour of orthoformate cryptands and reflects the low affinity of o-(H)2-1.1.1 for the sodium ion.
To explain the anomalous Li+/Na+ affinity of orthoformate cryptands (Chart 1e), a detailed analysis of the torsion angles (R–C–O–M dihedrals; R = H or CH3; M = Li or Na) was carried out. The average torsion angles in both orthoformate cryptands are significantly smaller with 168° for [Na+⊂o-(H)2-1.1.1] and 148° for [Li+⊂o-(H)2-1.1.1], when compared with the 180° found in [Na+⊂o-(CH3)2-1.1.1].9b The most striking result is the extremely narrow distribution of torsion angles around 180° in the orthoacetate cryptate, which was confirmed in at least three additional orthoacetate cryptands9c (see ESI†). Hence, the anti-relationship between the CH3 group and the Na+ ion seems to be strongly preferred in orthoacetate, but not orthoformate cryptands. This preference, which is likely due to a stereoelectronic effect,14 results in orthoacetate cages being effectively larger and more rigid than orthoformate cages and thus explains the observed cation selectivities.
In conclusion, we have described the template-free synthesis of o-(Hin)2-1.1.1, which to the best of our knowledge is the smallest organic cage compound15 prepared to date by reversible covalent reactions.16 X-ray crystallographic evidence tentatively supports our hypothesis that the process is thermodynamically driven by the formation of unusual intramolecular hydrogen bonds involving the bridgehead methine hydrogen atoms. Moreover, we demonstrated that bridgehead inversion occurs readily under conditions of dynamic covalent exchange.
This work was supported by the European Union (ERCstg 802428 “SUPRANET”) and the Deutsche Forschungsgemeinschaft (Emmy-Noether grant DE1830/2-1). We gratefully acknowledge Lanxess for a donation of Lewatit anion exchange resin. We thank Magdalena Heiland for experimental work carried out as part of her Master program and Bernhard Müller for assistance with X-ray crystallography.
Conflicts of interest
There are no conflicts to declare.Notes and references
- R. W. Alder and S. P. East, Chem. Rev., 1996, 96, 2097–2112 CrossRef CAS PubMed.
- (a) C. H. Park and H. E. Simmons, J. Am. Chem. Soc., 1968, 90, 2429–2431 CrossRef CAS; (b) J.-M. Lehn, Angew. Chem., Int. Ed. Engl., 1988, 27, 89–112 CrossRef; (c) P. G. Gassman and R. P. Thummel, J. Am. Chem. Soc., 1972, 94, 7183–7184 CrossRef CAS; (d) P. G. Gassman, S. R. Korn and R. P. Thummel, J. Am. Chem. Soc., 1974, 96, 6948–6955 CrossRef CAS.
- (a) C. H. Park and H. E. Simmons, J. Am. Chem. Soc., 1968, 90, 2431–2432 CrossRef CAS; (b) R. A. Bell, G. G. Christoph, F. R. Fronczek and R. E. Marsh, Science, 1975, 190, 151–152 CrossRef CAS; (c) S. O. Kang, J. M. Llinares, V. W. Day and K. Bowman-James, Chem. Soc. Rev., 2010, 39, 3980–4003 RSC.
- (a) P. B. Smith, J. L. Dye, J. Cheney and J.-M. Lehn, J. Am. Chem. Soc., 1981, 103, 6044–6048 CrossRef CAS; (b) B. Pool, S. Balalaie, A. Kunze, G. Schilling, P. Bischof and R. Gleiter, Eur. J. Org. Chem., 2004, 2812–2817 CrossRef CAS; (c) P. Morehouse, M. A. Hossain, J. M. Llinares, D. Powell and K. Bowman-James, Inorg. Chem., 2003, 42, 8131–8133 CrossRef CAS PubMed; (d) L. R. MacGillivray and J. L. Atwood, J. Org. Chem., 1995, 60, 4972–4973 CrossRef CAS; (e) L. R. MacGillivray and J. L. Atwood, Angew. Chem., Int. Ed. Engl., 1996, 35, 1828–1830 CrossRef CAS.
- (a) R. W. Alder, Tetrahedron, 1990, 46, 683–713 CrossRef CAS; (b) R. W. Alder and D. Read, Angew. Chem., Int. Ed., 2000, 39, 2879–2882 CrossRef CAS; (c) R. W. Alder, J. Chem. Soc., Perkin Trans. 2, 2001, 288–295 RSC.
- (a) M. Stollenz, N. Bhuvanesh, J. H. Reibenspies and J. A. Gladysz, Organometallics, 2011, 30, 6510–6513 CrossRef CAS; (b) M. Stollenz, D. Taher, N. Bhuvanesh, J. H. Reibenspies, Z. Baranová and J. A. Gladysz, Chem. Commun., 2015, 51, 16053–16056 RSC; (c) R. W. Alder, E. Honegger, A. B. Mcewen, R. E. Moss, E. Olefirowicz, P. A. Petillo, R. B. Sessions, G. R. Weisman, J. M. White and Z. Yang, J. Am. Chem. Soc., 1993, 115, 6580–6591 CrossRef CAS; (d) A. H. Haines and P. Karntiang, J. Chem. Soc., Perkin Trans. 1, 1979, 2577–2587 RSC; (e) M. Saunders and N. Krause, J. Am. Chem. Soc., 1990, 112, 1791–1795 CrossRef CAS; (f) R. S. Wareham, J. D. Kilburn, D. L. Turner, N. H. Rees and D. S. Holmes, Angew. Chem., Int. Ed. Engl., 1995, 34, 2660–2662 CrossRef CAS.
- (a) P. T. Corbett, J. Leclaire, L. Vial, K. R. West, J.-L. Wietor, J. K. M. Sanders and S. Otto, Chem. Rev., 2006, 106, 3652–3711 CrossRef CAS PubMed; (b) J. Li, P. Nowak and S. Otto, J. Am. Chem. Soc., 2013, 135, 9222–9239 CrossRef CAS PubMed; (c) Y. Jin, C. Yu, R. J. Denman and W. Zhang, Chem. Soc. Rev., 2013, 42, 6634–6654 RSC; (d) S. Otto, R. L. E. Furlan and J. K. M. Sanders, Curr. Opin. Chem. Biol., 2002, 6, 321–327 CrossRef CAS PubMed; (e) R. C. Brachvogel and M. von Delius, Eur. J. Org. Chem., 2016, 3662–3670 CrossRef CAS; (f) Q. Ji, R. C. Lirag and O. Š. Miljanić, Chem. Soc. Rev., 2014, 43, 1873–1884 RSC; (g) A. Herrmann, Chem. Soc. Rev., 2014, 43, 1899–1933 RSC; (h) A. Wilson, G. Gasparini and S. Matile, Chem. Soc. Rev., 2014, 43, 1948–1962 RSC.
- R.-C. Brachvogel and M. von Delius, Chem. Sci., 2015, 6, 1399–1403 RSC.
- (a) H. Löw, E. Mena-Osteritz and M. von Delius, Chem. Sci., 2018, 9, 4785–4793 RSC; (b) R.-C. Brachvogel, F. Hampel and M. von Delius, Nat. Commun., 2015, 6, 7129 CrossRef PubMed; (c) O. Shyshov, R. C. Brachvogel, T. Bachmann, R. Srikantharajah, D. Segets, F. Hampel, R. Puchta and M. von Delius, Angew. Chem., Int. Ed., 2017, 56, 776–781 CrossRef CAS PubMed; (d) X. Wang, O. Shyshov, M. Hanzevacki, C. M. Jäger and M. von Delius, J. Am. Chem. Soc., 2019, 141, 8868–8876 CrossRef CAS PubMed.
- R.-C. Brachvogel, H. Maid and M. von Delius, Int. J. Mol. Sci., 2015, 16, 20641–20656 CrossRef CAS PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- R. S. Rowland and R. Taylor, J. Phys. Chem., 1996, 100, 7384–7391 CrossRef CAS.
- R. D. Shannon, Acta Crystallogr., Sect. A: Found. Adv., 1976, 32, 751–767 CrossRef.
- I. V. Alabugin, Stereoelectronic Effects: A Bridge Between Structure and Reactivity, John Wiley & Sons Ltd, Chichester, 2016 Search PubMed.
- (a) J. Rodríguez, J. Mosquera, J. R. Couceiro, J. R. Nitschke, M. E. Vázquez and J. L. Mascareñas, J. Am. Chem. Soc., 2017, 139, 55–58 CrossRef PubMed; (b) M. Kolodziejski, A. R. Stefankiewicz and J.-M. Lehn, Chem. Sci., 2019, 10, 1836–1843 RSC; (c) W. Drozdz, C. Bouillon, C. Kotras, S. Richeter, M. Barboiu, S. Clément, A. R. Stefankiewicz and S. Ulrich, Chem. – Eur. J., 2017, 23, 18010–18018 CrossRef CAS PubMed.
- A. Maymounah and O. Š. Miljanić, Chem. Commun., 2018, 54, 11989–11997 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures, characterisation and spectral data, crystallographic data. CCDC 1876094 and 1876120. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9cc05968g |
| ‡ Removal of the metal from [Li+⊂o-(H)2-1.1.1] to o-(H)2-1.1.1 can be achieved quantitatively by treatment with anion exchange resin (Cl form, for further details see ESI†). |
| § Data fitted on the website http://www.supramolecular.org (95% confidence interval, original data is available for download, see ESI†). |
| This journal is © The Royal Society of Chemistry 2019 |