
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Transition metal-substituted Keggin polyoxotungstates enabling covalent attachment to proteinase K upon co-crystallization†
Joscha
Breibeck
 ,
Aleksandar
Bijelic
and
Annette
Rompel
*
,
Aleksandar
Bijelic
and
Annette
Rompel
*
Universität Wien, Fakultät für Chemie, Institut für Biophysikalische Chemie, Althanstraße 14, 1090 Wien, Austria. E-mail: annette.rompel@univie.ac.at; Web: http://www.bpc.univie.ac.at
First published on 6th September 2019
Abstract
The use of α- and β-Keggin polyoxotungstates (POTs) substituted by a single first row transition metal ion (CoII, NiII, CuII, ZnII) as superchaotropic crystallization additives led to covalent and non-covalent interactions with protein side-chains of proteinase K. Two major Keggin POT binding sites in proteinase K were identified, both stabilizing the orientation of the substituted metal site towards the protein surface and suggesting increased protein affinity for the substitution sites. The formation of all observed covalent bonds involves the same aspartate carboxylate, taking the role of a terminal oxygen with the Keggin α-isomer or even, in an unprecedented scenario, a bridging cluster oxygen with the β-isomer. Covalent bond formation with the protein carboxylate was observed only with the NiII- and CoII-substituted POTs, following the HSAB concept and the principle of metal immobilization.
Polyoxometalates (POMs) are anionic molecular oxo-clusters comprising early transition metals, in particular V, Mo and W, in high oxidation states. Recently, biological applications of POMs have been gaining attention1–3 and they were recognized for promising features aiding in protein crystallography.4 In addition to stabilizing crystal contacts, POMs provide strong anomalous signals as key to the phase problem.5 Thereby, they offer great potential for solving the X-ray structures of unknown proteins, as exploited in the famous example of the ribosomal subunits.6,7 In this case polyoxotungstates (POTs), and in about 30 others4 POM clusters were mainly introduced to the protein crystals via POM soaking. It is worth mentioning that derivatization of ribosomal proteins with small heavy atom ammine-complexes outperformed POTs in terms of structural resolution.8 However, when present in the crystallization drop from the start of the crystallization process, POTs can, besides acting as a phasing tool, promote the process of protein crystallization through the entropic gain of liberated hydration water9 and by facilitating new crystal contacts. Thereby, formation of novel crystal types as well as increased resolution can be achieved due to a more ordered protein packing and reduced flexibility, as proven using the Anderson–Evans hexatungstotellurate [TeW6O24]6− (TEW) as a protein crystallization additive.10
The success of TEW for protein crystallization served as an inspiration to investigate how the binding affinity of POTs towards proteins could be further increased beyond the interactions based on electronic polarization, purposefully aiming for the strongest interaction between two atoms: the covalent bond. A strong spatial fixation of POT clusters within the protein crystal by covalent attachment is highly advantageous to facilitate the localization of the anomalous W centers during anomalous phasing procedures, and crystal contacts can be stabilized more efficiently. To adapt the well-tried principle of metal immobilization11 for the requirements of protein crystallography on a molecular level, the chemically robust and monodisperse polyoxometalate scaffold could serve as a highly soluble chelating ligand suitable for co-crystallization with proteins.
In the present study, the Keggin archetypes α-[PW12O40]3−![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 12,13 and β-[SiW12O40]4−14,15 were chosen, which comprise a small POM scaffold allowing for substitution of outer shell addenda atoms (in this case: W) by transition metals,16 lanthanides17 and main group metal(loid)s.18 The spherical Keggin structure also provides an alternative scaffold geometry potentially preferring binding sites different from the planar Anderson structure. The incorporated transition metal is exposed and equipped with a labile water–ligand, which is susceptible to exchange by a more favorable ligand such as a nucleophilic protein side-chain. Proteinase K (from Tritirachium album)19 served as a well-characterized protein with a high ratio of positively charged side-chains (pI ≈ 8.920), which was expected to facilitate interactions with the POT polyanions (Fig. S1, ESI†).
12,13 and β-[SiW12O40]4−14,15 were chosen, which comprise a small POM scaffold allowing for substitution of outer shell addenda atoms (in this case: W) by transition metals,16 lanthanides17 and main group metal(loid)s.18 The spherical Keggin structure also provides an alternative scaffold geometry potentially preferring binding sites different from the planar Anderson structure. The incorporated transition metal is exposed and equipped with a labile water–ligand, which is susceptible to exchange by a more favorable ligand such as a nucleophilic protein side-chain. Proteinase K (from Tritirachium album)19 served as a well-characterized protein with a high ratio of positively charged side-chains (pI ≈ 8.920), which was expected to facilitate interactions with the POT polyanions (Fig. S1, ESI†).
The net charge of the mono-substituted Keggin clusters (Fig. S2, ESI†) stabilizes the resulting anions α-[PW11O39{M(H2O)}]5− (α-[PW11M]5−), here with M = CoII, NiII, CuII, ZnII, and β3-[SiW11O39{Co(H2O)}]6− (β-[SiW11Co]6−), against hydrolytic degradation from the acidic milieu up to the neutral pH range,21 which is compatible with protein crystallization in the pH range from 4 to 7. The five Keggin anions (see Fig. S2 and Table S1, ESI†) were synthesized according to literature procedures and experimentally confirmed by IR spectroscopy (Fig. S3 and Table S2, ESI†), TGA (Table S3, ESI†), elemental analysis (Table S4, ESI†) and ESI-MS (Table S5, ESI†). The solution stability of phosphate-centered POTs was additionally verified by 31P NMR22 (Fig. S4 and Table S6, ESI†), and protein stability in the presence of POMs was addressed by SDS-PAGE (Fig. S11, ESI†) and enzyme kinetics (Fig. S12, ESI†).
High resolution co-crystals of proteinase K and the Keggin anions (Table S7, ESI†) were obtained by hanging drop vapor diffusion in acetate buffer pH 5.5 for α-[PW11M]5− and in acetate buffer pH 4.5 with betaine for β-[SiW11Co]6−, using (NH4)2SO4 as a precipitant. Two main Keggin POT interaction sites were identified on the surface of proteinase K (termed position 1, Fig. 1, and position 2, Fig. 3). The polyanions substituted by NiII and CoII formed a strong covalent bond to the carboxylate side-chain of aspartate D207 (average metal–O distance: 1.7 Å, position 1, Fig. 1 and Fig. S6, ESI†) in vicinity to the conserved sulphate anion bound to R185. The observed 3d metal–oxygen distances for α-[PW11Co]5− and α-[PW11Ni]5− are slightly shorter than comparable distances reported before (Table S12, ESI†), possibly due to additional carboxylate π-interactions with the metal. Three protein chains participated in this interaction site (cf. Fig. S7A, ESI†). While the carboxylate oxygen of D207 replaced the water ligand at the transition metal within the α-Keggin structures (Fig. S2, ESI†), the β-isomeric structure resulted in the substitution of the adjacent μ2-O atom instead (Fig. 2), suggesting a hepta-coordinating W-site.23 In the Keggin β-isomer, β-[SiW11Co]6−, one {W3O12} triad is rotated by 60°, presenting the μ2-bridging POT oxygen atom for bond formation with D207. The μ2-O site linked to CoII is the most basic of the accessible oxygen atoms within the given POT framework24 and most likely to be intermediately protonated in the acidic environment applied, thereby facilitating the observed condensation with the protein side-chain. Such a μ2-O acylation reaction has been observed before with a self-activating (carboxyethyl)tin-substituted Wells–Dawson POT.25 Notably, the here-observed CoII–O bond length of 2.0 Å is consistent with the reported average range of 1.8–2.0 Å (Table S12, ESI†), suggesting a predominant σ-interaction of the carboxylate oxygen without significant π-participation. This supports the proposed binding scenario26,27 with a possible involvement of the non-bonding carboxylate π-system in binding to the tungsten center. The cluster β-[SiW11Co]6− was also engaged in another covalent bond formation to the carboxylate of D165 (Co–O distance: 1.8 Å, cf. Fig. S9, ESI†), mediating contacts to three protein molecules and emphasizing the strong propensity of CoII to bind carboxylate side-chains.
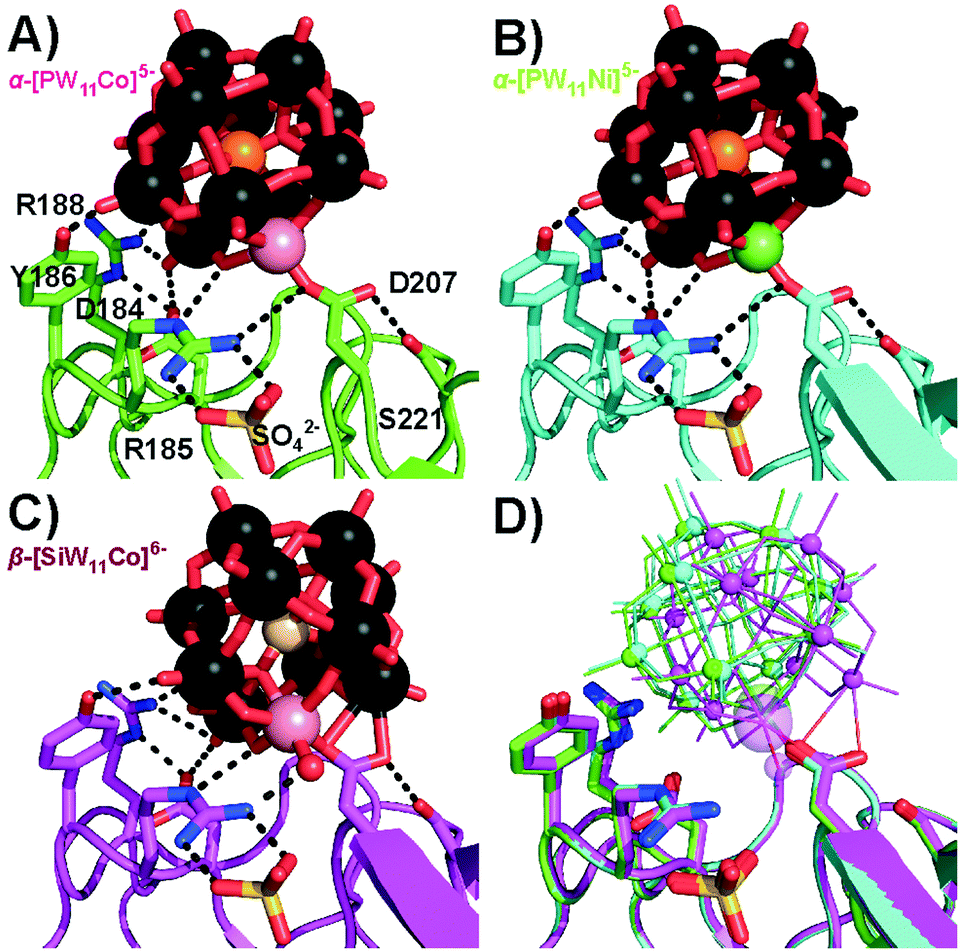 | ||
| Fig. 1 Covalent bond formation of Keggin POTs with D207 at position 1. R185 coordinating a sulphate anion is involved in strong hydrogen bonds to the POT oxygen atoms. Color code: W, black; O, red; P, orange; Si, ivory; Co, rose; Ni, green; N, blue; S, yellow. The interacting side-chains are depicted as sticks and interactions as black dashed lines (cf. Table S8 for bond lengths, ESI†). One-letter code for amino acids: D, aspartic acid; R, arginine; S, serine; Y, tyrosine. (A) α-[PW11Co]5−. (B) α-[PW11Ni]5−. (C) β-[SiW11Co]6−. The aquo-ligand is depicted as a red sphere and replaces the aspartate D207 carboxylate oxygen shown in (A) and (B) in H-bonding to arginine R185, while the other side-chain interactions are basically conserved. (D) Overlay of the three structures (A) (green), (B) (cyan), (C) (magenta) for comparison. Keggin POTs are shown in wire-frame mode with small spheres for W and large spheres for CoII or NiII. | ||
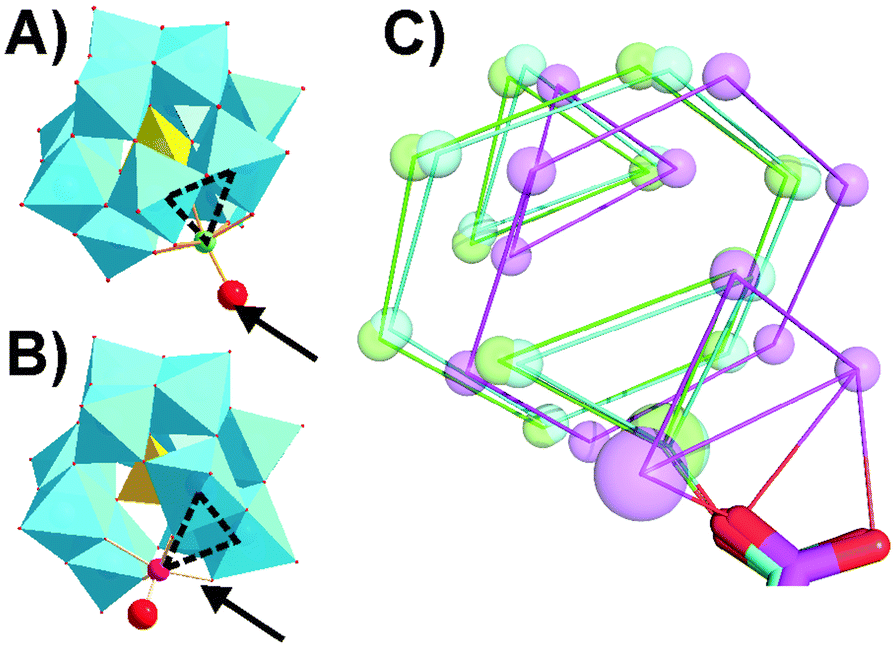 | ||
| Fig. 2 Keggin POT isomeric structures. (A) α-[PW11O39{M(H2O)}]5−. M = CoII, NiII. (B) β3-[SiW11O39{Co(H2O)}]6−. {WO6} units are depicted as cyan octahedra and the central atom as a yellow tetrahedron. The substituted transition metal M is indicated as a small sphere and the exchangeable aquo-ligand as a big red sphere. The dashed triangles indicate how the α- and β3-isomers of the Keggin structure differ by a 60° rotation of the respective W, W, M-triad, and the protein-conjugated positions found in the presented crystal structures are marked by arrows. (C) Schematic overlay of Keggin POT binding modes of the D207 carboxylate group to α-[PW11Co]5− (green), α-[PW11Ni]5− (cyan) and β-[SiW11Co]6− (magenta). W atoms are shown as small spheres and CoII or NiII as large spheres. Metal atoms within a common plane are connected for direct structural comparison. | ||
Interestingly, α-[PW11Cu]5− and α-[PW11Zn]5− were not observed at position 1, but shared the second main position close to residue S45 with the other three Keggin anions. In this position 2 (Fig. 3), no covalent bond between the protein and POT cluster was formed. All five Keggin clusters are rather coordinated by hydrogen bonds, mainly to the main-chain peptide backbone, and the aquo-ligand at the transition metal atom plays an important role to interconnect the POT anions with the surface hydration water molecules of the protein. This water ligand is not only an H-acceptor, but also an H-donor, which makes it a more flexible H-bonding moiety than the terminal H-accepting oxo-ligand usually present in unsubstituted Keggin anions. The five Keggin POTs at position 2 interact with two protein molecules within the crystal (cf. Fig. S7B, ESI†). The structural isomer β-[SiW11Co]6− adopted a slightly tilted orientation at position 2 (Fig. 3) when compared to the α-isomers to accommodate the water ligand at the very same position as found for those structures. The CuII- and ZnII-substituted POT anions do not carry an aquo-ligand at the transition metal center based on our X-ray data, most likely due to their peculiar coordination chemistry and in line with the lack of observation of a covalent bond in position 1. The resulting reduced binding stability of these two compounds was reflected in overall lower POT occupancies as well as a reduced spatial fixation at position 2 (cf. Fig. S10 and Table S10, ESI†). With no preferential orientation and the transition metal site pointing away from the protein surface, α-[PW11Cu]5− described a pathway sliding through the solvent channels in between the protein molecules.
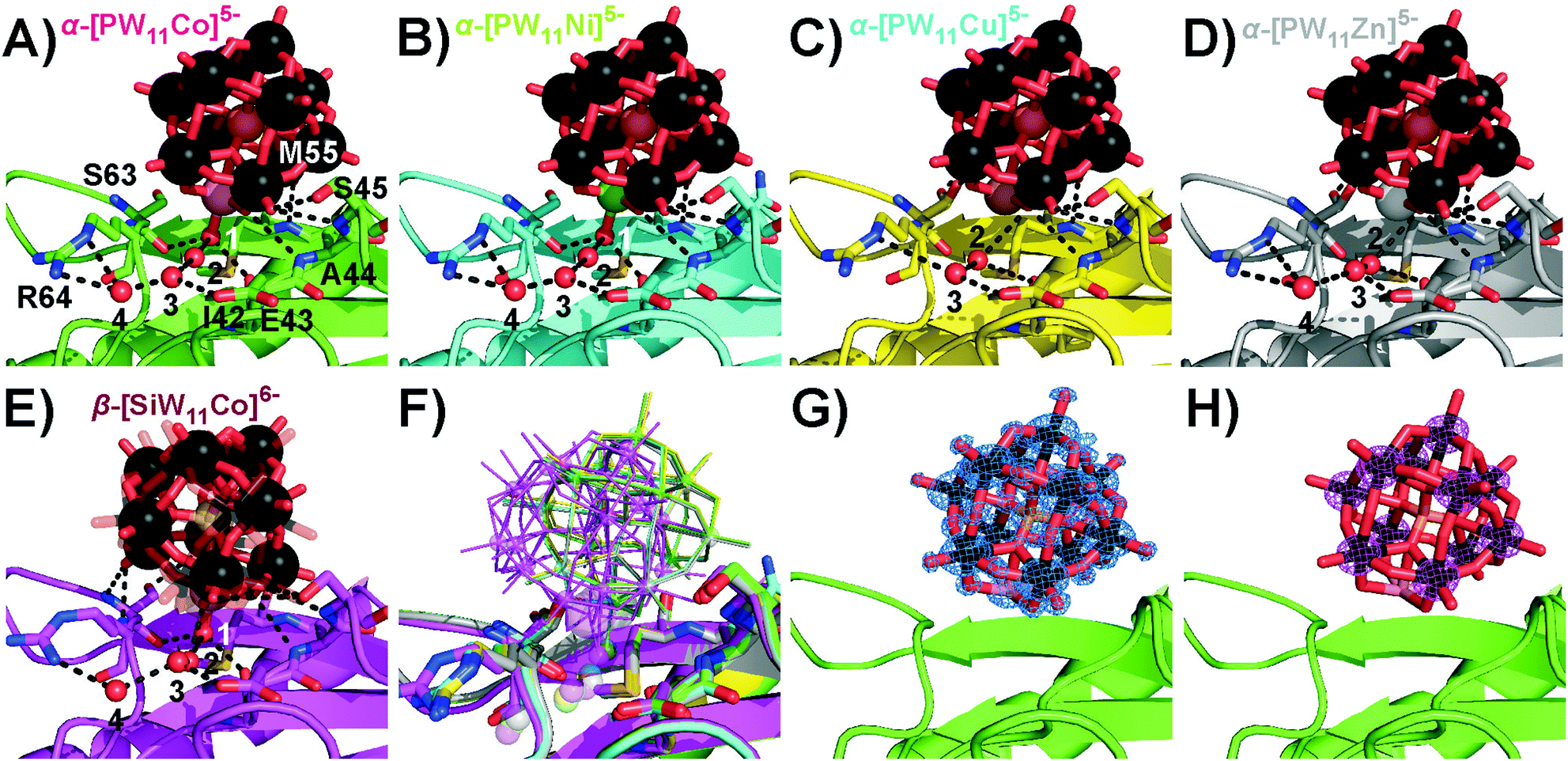 | ||
| Fig. 3 Participation of the Keggin-POT aquo-ligand in the hydrogen network (black dashed lines, cf. Table S9, ESI† for bond lengths) in the vicinity of S45 in position 2. The aquo-ligand is assigned as number 1, and other conserved crystal water molecules are numbered by 2 to 4. While mostly side-chains stabilize the protein–POT interactions around D207, amide H-bonding by the main chain is the main contribution found for the present POT interaction site. One-letter code for amino acids: A, alanine; E, glutamic acid; I, isoleucine; M, methionine; R, arginine; S, serine. (A) α-[PW11Co]5−. (B) α-[PW11Ni]5−. (C) α-[PW11Cu]5−. (D) α-[PW11Zn]5−. No electron density for an aquo-ligand was found close to the copper or zinc atom, suggesting a reduced electronic availability of the CuII or ZnII center for an additional ligand when compared to CoII and NiII. The concomitant missing stabilization by H-bonding results in a strongly reduced occupancy and spatial fixation of the Keggin-POT in this position (cf. Table S10, ESI†). (E) β-[SiW11Co]6−. Two slightly shifted positions were found for this POT, which both are kept in place by the water ligand retaining its common position known from structures (A) and (B). One position is depicted with increased transparency for the sake of clarity. (F) Overlay of selected structures (A–E) for comparison. Transition metals are indicated as small (W) or big spheres (Co, Ni, Cu, Zn). (G) α-[PW11Co]5− model with 2Fc–Fo electron density map. All cluster atoms are defined to high resolution. (H) Anomalous difference map for the same cluster. The 11 tungsten atoms give rise to substantial anomalous density. | ||
The observed POT binding modes and affinities to the protein side-chains were further analyzed based on the following considerations. The basic nitrogen-bearing side-chains were fully protonated in the acidic crystallization milieu applied (pH 4.5–5.5), and the carboxylate groups presented the strongest available nucleophiles to bind to the exposed transition metals.
As the hardness of the metal(II) centers decreases within the series CoII > NiII > CuII > ZnII following the HSAB concept, their tendency to bind hard oxygen ligands such as carboxylates decreases.28 In the same direction the preferred coordination geometry changes from octahedral to square planar, with increasing distortions due to the electronic configurations.29 As CuII and ZnII are electronically saturated within the pentadentate square pyramidal binding site of the Keggin POT lacuna,30 they do not necessarily require a sixth ligand, so they hardly bind to protein side-chains in the given chemical environment (cf. Table S11, ESI†). In the crystal structures, the electron density of the Keggin POT anions was well-defined down to atomic scale (Fig. 3G), and the strong anomalous signal of the tungsten atoms (Fig. 3H) facilitated proper placement and orientation of the cluster structures in the model after molecular replacement (Table S7, ESI†).
The first covalent attachment of a polyoxometalate cluster used as a crystallization additive was observed with the POT TEW,31 which adopted a bent structure to accommodate the carboxylate oxygen atoms of an aspartate side-chain. The bond formation was presumably forced by the strong electrostatic interactions of TEW in the positively charged environment of the binding-site, and did not arise from a pronounced affinity of the saturated W atoms to the side-chain. The metal–O distances in this weak bond were considerably longer (2.4 Å) than the majority of bond lengths observed in this study (1.5–1.9 Å). Recently, a Zr-containing Keggin anion was co-crystallized with hen egg-white lysozyme leading to a linkage of the cluster with a glutamine carboxamide,32 a presumable complex intermediate previous to carboxamide hydrolysis triggered by the strongly Lewis-acidic ZrIV center. However, co-crystallizations of another transition-metal substituted POT of the Wells–Dawson archetype with lysozyme yielded only non-covalent complexes with the protein, and the substitution sites faced away from the protein surface with no preferential orientation.33 In contrast, the Keggin POTs in this study surpassed mere electrostatic interactions with the protein surface.
These novel crystallization additives can be expected to stabilize flexible protein regions and to support strong crystal lattice interactions. The broad pH stability range of the selected POT anions facilitated their application as crystallization additives and phasing tools. Directed bond formation with proteins marks the next step in the development of universal POT crystallization additives for covalent cross-linking of protein molecules, and also stimulates further investigations of POT–protein interactions in other fields.
Experimental details can be found in the detailed SI document available online. The crystal structures obtained in this study are available from the Protein Data Bank (http://www.rcsb.org) as PDB entries 6RUG, 6RUH, 6RUK, 6RUN, 6RUW, 6RVE and 6RVG.
This work was supported by funding by the Austrian Science Fund (FWF): P27534 and the University of Vienna. We thank the staff at ESRF, and of EMBL Grenoble, especially Dr Ulrich Zander, for assistance and support during the beamtime at beamline ID23-1 (MX-1831) allocated to the Austrian Crystallographic Diffraction Consortium (AC-DC).
Conflicts of interest
There are no conflicts to declare.References
- A. Bijelic, M. Aureliano and A. Rompel, Chem. Commun., 2018, 54, 1153–1169 RSC.
- A. Bijelic, M. Aureliano and A. Rompel, Angew. Chem., Int. Ed., 2019, 58, 2980–2999 ( Angew. Chem. , 2019 , 131 , 3008–3029 ) CrossRef CAS.
- M. Arefian, M. Mirzaei, H. Eshtiagh-Hosseini and A. Frontera, Dalton Trans., 2017, 46, 6812–6829 RSC.
- A. Bijelic and A. Rompel, Coord. Chem. Rev., 2015, 299, 22–38 CrossRef CAS.
- A. Bijelic and A. Rompel, ChemTexts, 2018, 4, 10 CrossRef.
- F. Schluenzen, A. Tocilj, R. Zarivach, J. Harms, M. Gluehmann, D. Janell, A. Bashan, H. Bartels, I. Agmon, F. Franceschi and A. Yonath, Cell, 2000, 102, 615–623 CrossRef CAS.
- J. Harms, F. Schluenzen, R. Zarivach, A. Bashan, S. Gat, I. Agmon, H. Bartels, F. Franceschi and A. Yonath, Cell, 2001, 107, 679–688 CrossRef CAS.
- N. Ban, P. Nissen, J. Hansen, P. B. Moore and T. A. Steitz, Science, 2000, 289, 905–920 CrossRef CAS.
- C. Molitor, A. Bijelic and A. Rompel, IUCrJ, 2017, 4, 734–740 CrossRef CAS.
- A. Bijelic and A. Rompel, Acc. Chem. Res., 2017, 50, 1441–1448 CrossRef CAS.
- J. Porath, Protein Expression Purif., 1992, 3, 263–281 CrossRef CAS.
- J. C. Bailar, in Inorganic Syntheses, ed. H. S. Booth, The McGraw-Hill Book Company, Inc., New York, 1939, vol. 1, pp. 132–133 Search PubMed.
- J. F. Keggin, Nature, 1933, 131, 351 CrossRef.
- A. Tézé, G. Hervé, R. G. Finke and D. K. Lyon, in Inorganic Syntheses, ed. A. P. Ginsberg, John Wiley & Sons, Inc., Hoboken, 1990, vol. 27, pp. 85–96 Search PubMed.
- J. Fuchs, A. Thiele and R. Palm, Z. Naturforsch. B, 1981, 36, 161–171 Search PubMed.
- T. J. R. Weakley and S. A. Malik, J. Inorg. Nucl. Chem., 1967, 29, 2935–2944 CrossRef CAS.
- B. S. Bassil, M. H. Dickman, B. von der Kammer and U. Kortz, Inorg. Chem., 2007, 46, 72452–72458 CrossRef.
- P. Gouzerh and A. Proust, Chem. Rev., 1998, 98, 77–112 CrossRef CAS.
- W. Saenger in Handbook of Proteolytic Enzymes, ed. N. D. Rawlings and G. Salvesen, Academic Press, New York, 2013, vol. 3, pp. 3240–3242 Search PubMed.
- W. Ebeling, N. Hennrich, M. Klockow, H. Metz, H. D. Orth and H. Lang, Eur. J. Biochem., 1974, 47, 91–97 CrossRef CAS.
- J. Breibeck, N. I. Gumerova, B. B. Boesen, M. Galanski and A. Rompel, Sci. Rep., 2019, 9, 5183 CrossRef.
- R. I. Maksimovskaya and G. M. Maksimov, Coord. Chem. Rev., 2019, 385, 81–99 CrossRef CAS.
- J. Gao, J. Yan, S. Beeg, D. L. Long and L. Cronin, J. Am. Chem. Soc., 2013, 135, 1796–1805 CrossRef CAS.
- X. López, C. Bo and J. M. Poblet, J. Am. Chem. Soc., 2002, 124, 12574–12582 CrossRef.
- C. Boglio, K. Micoine, É. Derat, R. Thouvenot, B. Hasenknopf, S. Thorimbert, E. Lacôte and M. Malacria, J. Am. Chem. Soc., 2008, 130, 4553–4561 CrossRef CAS.
- B. Burger, S. Dechert, C. Große, S. Demeshko and F. Meyer, Chem. Commun., 2011, 47, 10428–10430 RSC.
- M. A. Gil, W. Maringgele, S. Dechert and F. Meyer, Z. Anorg. Allg. Chem., 2007, 633, 2178–2186 CrossRef CAS.
- L. Rulíšek and J. Vondrášek, J. Inorg. Biochem., 1998, 71, 115–127 CrossRef.
- D. Venkataraman, Y. Du, S. R. Wilson, K. A. Hirsch, P. Zhang and J. S. Moore, J. Chem. Educ., 1997, 74, 915–918 CrossRef CAS.
- J. A. Gamelas, I. C. M. S. Santos, C. Freire, B. de Castro and A. M. V. Cavaleiro, Polyhedron, 1999, 18, 1163–1169 CrossRef CAS.
- C. Molitor, A. Bijelic and A. Rompel, Chem. Commun., 2016, 52, 12286–12289 RSC.
- L. Vandebroek, L. van Meervelt and T. N. Parac-Vogt, Acta Crystallogr., Sect. C: Struct. Chem., 2018, 74, 1348–1354 CrossRef CAS.
- L. Vandebroek, Y. Mampaey, S. Antonyuk, L. van Meervelt and T. N. Parac-Vogt, Eur. J. Inorg. Chem., 2018, 506–511 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9cc05818d |
| This journal is © The Royal Society of Chemistry 2019 |