
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNeutralization of insulin by photocleavage under high vacuum†
Jonas
Schätti
a,
Moritz
Kriegleder
b,
Maxime
Debiossac
 b,
Michael
Kerschbaum
b,
Philipp
Geyer
b,
Marcel
Mayor
acd,
Markus
Arndt
*b and
Valentin
Köhler
*a
b,
Michael
Kerschbaum
b,
Philipp
Geyer
b,
Marcel
Mayor
acd,
Markus
Arndt
*b and
Valentin
Köhler
*a
aDepartment of Chemistry, University of Basel, St. Johannsring 19, CH Basel, Switzerland. E-mail: valentin.koehler@unibas.ch
bFaculty of Physics, University of Vienna, Boltzmanngasse 5, A-1090 Vienna, Austria. E-mail: markus.arndt@univie.ac.at
cInstitute of Nanotechnology, Karlsruhe Institute of Technology, Hermann-von-Helmholtz-Platz 1, 76344 Eggenstein-Leopoldshafen, Germany
dLehn Institute of Functional Materials (LIFM), Sun Yat-Sen University (SYSU), XinGangXi Road 135, 510275 Guangzhou, P. R. China
First published on 25th September 2019
Abstract
Charge reduction and neutralization of electro-sprayed peptides are realized by selective gas-phase photocleavage of tailored covalent tags. The concept is demonstrated with four model peptides in positive and negative ion modes and tagged insulin as the largest construct.
Already thirty years ago, electrospray ionization, ESI,1 and matrix-assisted laser desorption ionization, MALDI,2,3 were established as efficient ways of transferring charged macromolecules into the gas phase for mass spectrometry. ESI excels in producing continuous beams of highly charged and even massive biomolecules under mild conditions and it is readily coupled with on-line separation methods, such as high-performance liquid chromatography. MALDI, on the other hand, transfers short pulses of mostly singly charged macromolecules into the gas phase. Both techniques complement each other and have become essential in biomolecular analysis. A high charge-per-mass value favours a high velocity in time-of-flight mass spectrometry (TOF-MS) and thus boosts the detection efficiency for heavy molecules. On the other hand, a low charge value and a narrow charge distribution simplify the mass spectrum of complex molecular mixtures.4
Even though MALDI5 produces a substantial fraction of neutral material, mostly trapped in nanoparticles or droplets, the controlled preparation of neutral mass-selected peptide or protein beams has remained a challenge, up to now. If this technology becomes available, it is expected to open a new window to electric and magnetic beam deflection,6,7 action-free single-photon recoil spectroscopy8,9 as well as a new branch of molecular quantum optics with proteins.10 While aqueous solutions are the normal habitat of biomolecules in nature, gas phase studies on isolated specimens of selectable charge states, including neutrals, will add to our overall understanding of the electronic and structural properties of this important class of organic molecules.11
The goal of our present work is to establish a chemical and optical strategy to volatilize complex biopolymers, to control their motion and their charge at a given time and position using photo-induced cleavage of charged tags. Photocleavage itself is an established technique, recently studied in a variety of experiments on trapped ions, for instance to prepare diagnostic fragments for molecular structure determination.12–14 Signal depletion of laser desorbed, non-peptidic model compounds was observed after irradiation.15 Similar observations were made with peptides, launched by ESI where additionally clean cleavage was confirmed.16
Here we introduce photocleavable tags with charge-decorated leaving groups for peptides and proteins. They can be removed in an overall non-heterolytic process that reduces the peptide charge stepwise by one unit:
| (X–negLG)−N → X−N+1 + negLG− as well as (X–posLG)+N → X+N−1 + posLG+ |
In contrast to that, earlier studies found that a similarly structured neutral leaving group neuLG cleaves heterolytically for tripeptides and mostly via H-transfer for longer peptides, thereby limiting the applicability for charge reduction by photocleavage to small peptides in the negative ion mode (Scheme 1).16
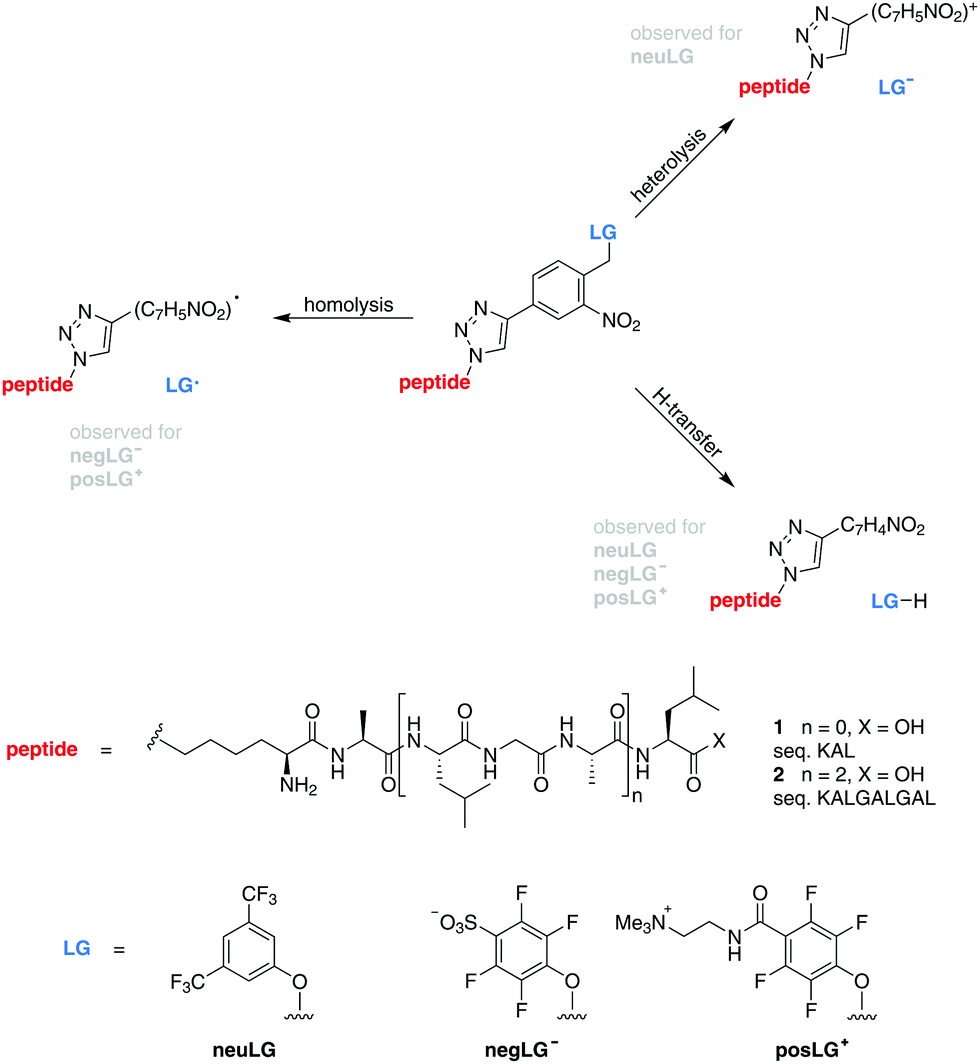 | ||
| Scheme 1 Model peptides for charge-controlled photocleavage. Tripeptides 1 (n = 0) and nonapeptides 2 (n = 2) are modified by an ortho-nitroarylether group17,18 connected to a neutral (neuLG), negative (negLG−) or positive (posLG+) leaving group. | ||
In our present work, fluorinated phenols with either a positive tetraalkylammonium (posLG+) or a negative sulfonate (negLG−) group are explored as leaving group motifs.15,16 They are bound to a set of tri- and nonapeptides 1 and 2 (sequences: KAL and KALGALGAL, respectively) shown in Scheme 1, which contain only non-aromatic amino acids to minimize absorption. Because of this choice, the phototag is the strongest absorber in the molecule at 266 nm.
The photocleavable group is based on the well-established ortho-nitrobenzyl ether motif.17,18 Although there is only limited information on its gas-phase cleavage behaviour available, it seems reasonable to assume that the cleavage under H-transfer results in the formation of a nitrosobenzaldehyde moiety as is observed in solution. The homolytic and heterolytic pathways might likewise proceed via a cyclic intermediate. Further studies are however required to elucidate the underlying mechanisms of the observed processes.
The peptides are volatilized using micro-electrospray in a Waters Q-TOF mass spectrometer, which we upgraded using a high voltage corona discharge chamber to reduce the peptide charge in collision with bi-polar air.19 The molecules are then guided into high vacuum where they are selected using a quadrupole mass filter, which was upgraded for high mass-to-charge states, up to 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 u e−1. The ions pass on to a hexapole guide for collisional cooling and/or dissociation in a dilute argon buffer gas. A beam of short-pulse (10 ns) ultraviolet (266 nm) laser light is aligned collinearly with the ion beam to induce the photocleavage of the peptides when they leave the collision cell. The laser was kept at a pulse rate adjusted to the speed of the molecules (see the ESI†) and at an intensity of 0.5–1 mW cm−2. The parent ions and their cleavage products are then analysed by high-resolution time-of-flight mass spectrometry.16
000 u e−1. The ions pass on to a hexapole guide for collisional cooling and/or dissociation in a dilute argon buffer gas. A beam of short-pulse (10 ns) ultraviolet (266 nm) laser light is aligned collinearly with the ion beam to induce the photocleavage of the peptides when they leave the collision cell. The laser was kept at a pulse rate adjusted to the speed of the molecules (see the ESI†) and at an intensity of 0.5–1 mW cm−2. The parent ions and their cleavage products are then analysed by high-resolution time-of-flight mass spectrometry.16
The data of Fig. 1 show that our model compounds fulfill their design specifications well: the charged leaving groups negLG− and posLG+ cleave under H-transfer or homolytically, without resulting in charge transfer between the dissociating partners. Both processes reduce the charge of the peptide moiety in a controlled fashion.
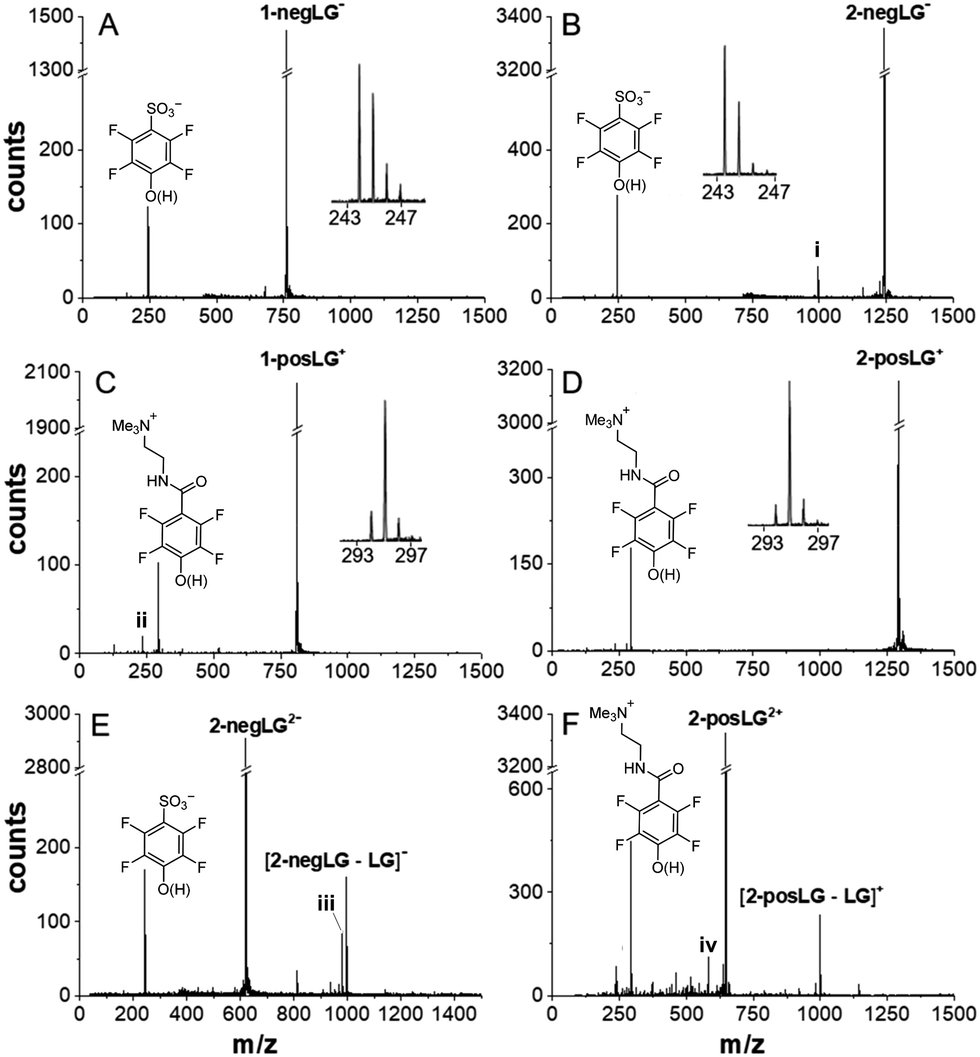 | ||
| Fig. 1 Photocleavage pathways for peptides with charged leaving groups. The peptides were initially mass selected to the single parent peak. All additional peaks are caused by the interaction with the laser light. Cleavage of negative model peptides (A) 1-negLG− and (B) 2-negLG−, positive model peptides (C) 1-posLG+ and (D) 2-posLG+, and doubly charged peptides (E) 2-negLG2− and (F) 2-posLG2+ takes place. All insets identify the leaving groups and allow estimation of the fraction of cleavage under homolysis and H-transfer. A signal at 996 amu (i) in panel (B) indicates a peptide whose charge is located on the peptide fragment rather than the leaving group. Signals (ii) and (iv) in (C) and (F) indicate loss of NMe3 from the leaving group. The signal (iii) indicates the additional formation of a 16 amu lighter fragment as observed previously.16 The vertical figure axes were cut to facilitate a comparison of the peaks. | ||
Cleavage of both the negative tripeptide 1-negLG− and the nonapeptide 2-negLG− results in a clean mass spectrum, containing the parent and the leaving group (Fig. 1(A) and (B)), but no or little of the mass-reduced peptide moiety. This corroborates that the negative charge stays on the leaving group and the neutral peptide remains undetected. A comparison of the mass peaks shows that homolysis (h) and H-transfer (H) contribute with a ratio between h/H ≃ 2:1 and 1:1 to the dissociation process.
In contrast, the positive ions 1-posLG+ and 2-posLG+ cleave preferentially under H-transfer, with a ratio of h/H = 1:5 to 1:10. Heterolytic cleavage into a neutral posLG− and a positive peptide was never observed in our studies. Again, the absence of charged peptide fragments in Fig. 1(C) and (D) indicates that they are neutral. To corroborate this statement and to test for possibly competing fragmentation pathways we performed experiments on the doubly charged nonapeptides 2-negLG2− and 2-posLG2+. They hold one charge on the leaving group and one on the peptide moiety and both the LG fragments and the peptide are observed as singly charged peaks in Fig. 1(E) and (F). Even though homolytic cleavage generates radicals, we do not observe any excessive fragmentation. By analogy, we assume that neutral peptides generated in a photocleavage process will not further dissociate either.
While an earlier study using neutral tags reported that the cleavage mechanism can change with peptide length, all phototags of our present study consistently cleave without charge transfer for tripeptides as well as for nonapeptides. It is therefore intriguing to explore whether our new tags allow a controlled charge reduction of even large polypeptides or proteins. This has motivated our study on insulin, tagged with three negative leaving groups, here termed negIns. The tags were introduced by reactions with the corresponding NHS-esters and are assumed to be located at the two N-termini and the single lysine residue (see the ESI†).
Insulin contains seven aromatic amino acids, four tyrosine and three phenylalanine residues, which compete with the phototags for UV absorption. However, their combined extinction coefficient in solution amounts to ε ∼ 4200 M−1 cm−1 and is small in comparison with the combined extinction coefficient of all nitroaryl groups εnitrobenzene ∼ 20000 M−1 cm−1.20,21
The desired initial charge state is optimized by guiding the electrosprayed insulin through bipolar air in a high-voltage DC discharge.19 Subsequent high-pass mass filtering before the molecule-laser interaction ensures that all subsequently detected low-mass molecules are due to photocleavage.
In Fig. 2 we show the photo response of negIns for the initial charge states q = −7 to q = −2.
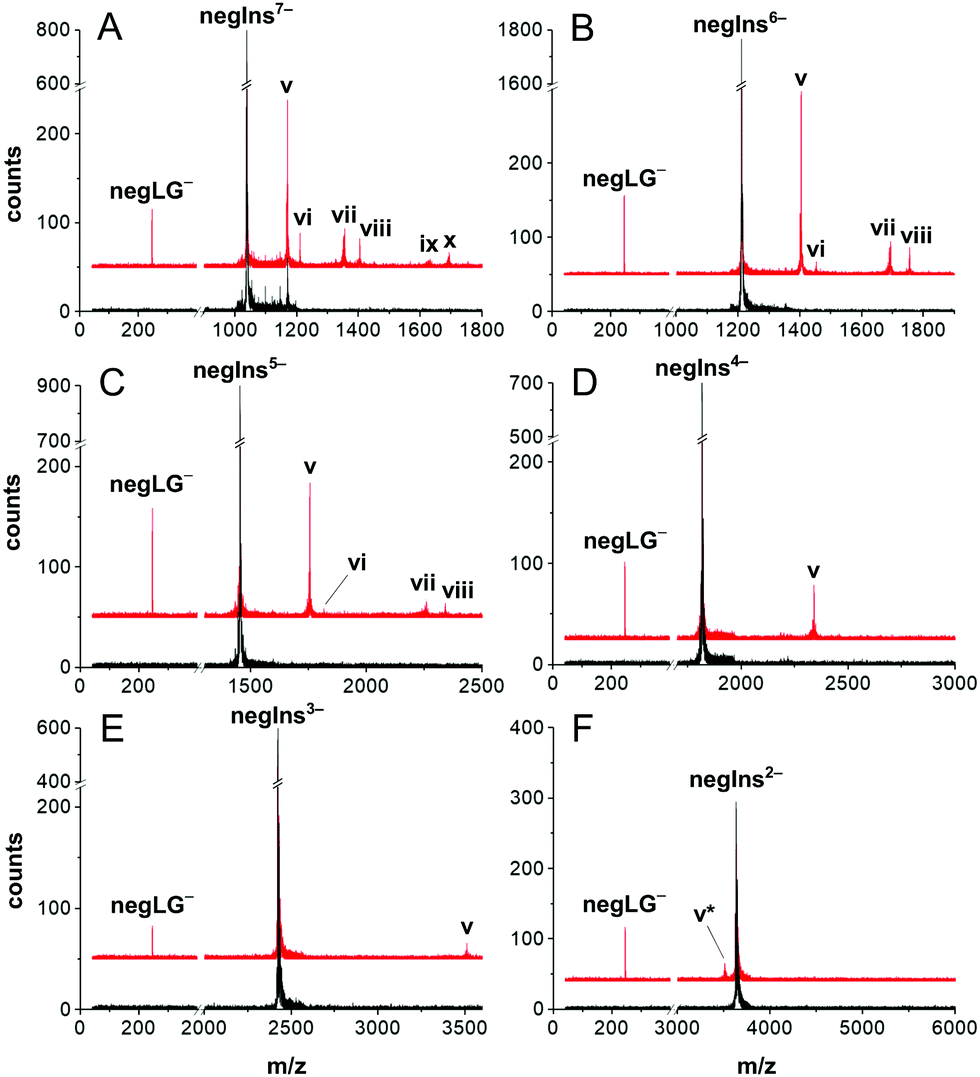 | ||
| Fig. 2 Charge reduction of an insulin ion beam by controlled photocleavage. NegIns dissociates in defined fragments under irradiation by 266 nm light. The panels (A–F) represent different initial charge states from q = −7 down to q = −2. In all charge states we can observe similar processes. (v) The charge of the parent molecule M−N is reduced by the loss of negLG− (M-LG)−N+1 and thus appears at higher m/z values. (vi) Electron detachment (M-e)−N+1. (vii) Two leaving groups (M-2LG)−N+2 are removed from highly charged insulin M7−–M5−. (viii) Cleavage and detachment can occur together (M-LG-e)−N+2. (ix) A tiny amount of (M7−) molecules even splits off three charged leaving groups to yield (M-3LG)−N+3. (x) (M-2LG-e)−N+3. (v*) The loss of negLG with a protonated sulfonate is observed in small amounts (M-LG)−N. Black lines: mass spectra without laser irradiation. Red line: mass spectra with laser irradiation, upshifted by 50 counts for clarity. | ||
In panel (A) we observe photocleavage of a singly charged leaving group (peak v), electron photo-detachment (vi), cleavage of two charged leaving groups (M-2LG)5− (vii) as well as mixed processes, such as the cleavage of one tag and the emission of an electron resulting in (M-LG-e)5− (viii) and photocleavage of all leaving groups (M-3LG)4− (ix). Most processes, listed for the 7-fold charged anion, also occur in less highly charged molecules but decrease with molecular charge and become irrelevant below q = −4. This is consistent with earlier photodetachment studies of insulin in ion traps.22
The experimental results confirm the design purpose of our molecular tags: for insulin in a low initial charge state, controlled photocleavage is the only relevant charge reduction process.
Photodepletion of the parent ion peak – here defined as 1 − M/M0 where M/M0 is the ion signal with/without laser irradiation – attained values as high as 30% on the doubly charged insulin anion and 40–60% for insulin with charge states q = −4 to q = −7 (Fig. 3B).
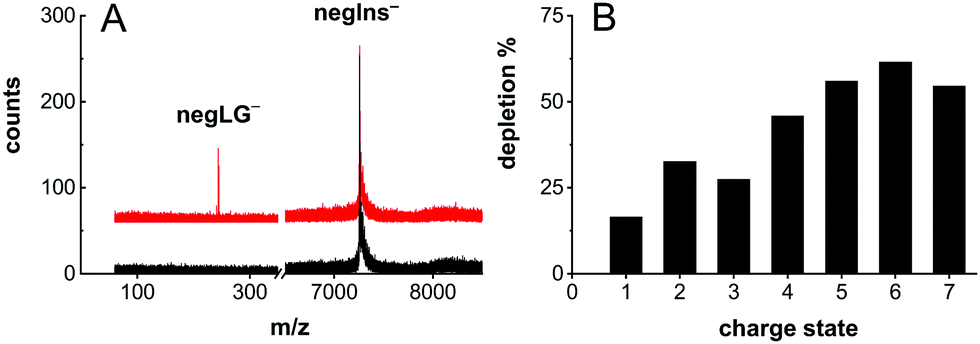 | ||
| Fig. 3 Photocleavage neutralization of negIns. (A) The mass spectrum of negIns− with (red) and without (black) laser irradiation. The appearance of negLG− upon laser irradiation indicates the formation of neutral insulin (negIns-LG)0. (B) The depletion efficiency decreases as a function of the charge state. | ||
Photocleavage even enables efficient neutralization of insulin (Fig. 3A).
This is shown in Fig. 3A, where the triply tagged, singly charged molecule is neutralized by removing a singly charged leaving group.
The remaining insulin is still decorated with 2 PCTs and a small PCT fragment. One may expect that such small tags, similar in size to fluorescent labels used in biology, will influence the molecular structure only little.
Summarizing, we have designed and synthesized a new set of charged molecular tags that allow one to control the photocleavage of peptides generically, from small tripeptides up to a complex biopolymer as massive as insulin with 51 amino acid residues. Contrary to earlier observations with neutral tags, where the cleavage mechanism varied with peptide size, the charged tags used in our present study enable efficient photocleavage charge reduction over a wide scale of molecular mass and over many amino acid residues. We have proven this for the mass range of 1–7 kDa and for charge states from one to seven. To our knowledge, this is the first evidence for controlled photocleavage neutralization of a high-mass polypeptide such as human insulin under high vacuum. First experiments for charge reduction with an analogous positively charged insulin construct (posIns) suggest that the methodology can be equally applied in a positive ion mode (see the ESI†).
Future studies will explore the reionization of neutral proteins via a second photocleavable tag tailored for a second wavelength. The efficiency of the process will be substantially enhanced using laser light in a cross-beam enhancement cavity or using intense ultrafast laser pulses with high repetition rates. By appropriate design of the photo-cleavable chromophore it will be possible to choose a laser wavelength beyond the natural absorption profile of the biopolymer. This shall become the basis for preparing continuous cold beams of mass-selected neutral proteins for UV/VIS/IR spectroscopy, deflectometry and quantum interferometry as well as new techniques for photo-cleavage assisted mass spectrometry.
JS and VK are grateful for continuous support and lab space from T. R. Ward (University of Basel). We thank Simone Auer for experimental support. This project has received funding from the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation program (MA, Grant No. 320694) and the Austrian Science Fund (FWF) within program W1210-N25. The synthesis was supported by the Swiss National Science Foundation (200020-178808) and the Swiss Nanoscience Institute (P1403). MM gratefully acknowledges support from the 111 project (90002-18011002).
Conflicts of interest
There are no conflicts to declare.Notes and references
- J. B. Fenn, M. Mann, C. K. Meng, S. F. Wong and C. M. Whitehouse, Science, 1989, 246, 64–71 CrossRef CAS PubMed.
- M. Karas and F. Hillenkamp, Anal. Chem., 1988, 60, 2299–2301 CrossRef CAS PubMed.
- K. Tanaka, H. Waki, Y. Ido, S. Akita, Y. Yoshida and T. Yoshida, Rapid Commun. Mass Spectrom., 1988, 2, 151–153 CrossRef CAS.
- L. M. Smith, J. Am. Soc. Mass Spectrom, 2008, 19, 629–631 CrossRef CAS PubMed.
- R. Knochenmuss and R. Zenobi, Chem. Rev., 2003, 103, 441–452 CrossRef CAS PubMed.
- R. Antoine and P. Dugourd, Phys. Chem. Chem. Phys., 2011, 13, 16494–16509 RSC.
- R. Antoine, I. Compagnon, D. Rayane, M. Broyer, P. Dugourd, N. Sommerer, M. Rossignol, D. Pippen, F. C. Hagemeister and M. F. Jarrold, Anal. Chem., 2003, 75, 5512–5516 CrossRef CAS PubMed.
- S. Eibenberger, X. Cheng, J. P. Cotter and M. Arndt, Phys. Rev. Lett., 2014, 112, 250402 CrossRef PubMed.
- J. Rodewald, P. Haslinger, N. Dörre, B. A. Stickler, A. Shayeghi, K. Hornberger and M. Arndt, Appl. Phys. B: Lasers Opt., 2017, 123, 3 CrossRef PubMed.
- M. Arndt, Phys. Today, 2014, 67, 30–36 CrossRef.
- M. F. Jarrold, Annu. Rev. Phys. Chem., 2000, 51, 179–207 CrossRef CAS.
- M. W. Forbes, A. M. Nagy and R. A. Jockusch, Int. J. Mass Spectrom., 2011, 308, 155–166 CrossRef CAS.
- N. G. Hendricks, N. M. Lareau, S. M. Stow, J. A. McLean and R. R. Julian, J. Am. Chem. Soc., 2014, 136, 13363–13370 CrossRef CAS PubMed.
- F. Turecek and R. R. Julian, Chem. Rev., 2013, 113, 6691–6733 CrossRef CAS PubMed.
- U. Sezer, P. Geyer, M. Kriegleder, M. Debiossac, A. Shayeghi, M. Arndt, L. Felix and M. Mayor, Beilstein J. Nanotechnol., 2017, 8, 325–333 CrossRef CAS PubMed.
- M. Debiossac, J. Schätti, M. Kriegleder, P. Geyer, A. Shayeghi, M. Mayor, M. Arndt and V. Köhler, Phys. Chem. Chem. Phys., 2018, 20, 11412–11417 RSC.
- P. Klán, T. Šolomek, C. G. Bochet, A. Blanc, R. Givens, M. Rubina, V. Popik, A. Kostikov and J. Wirz, Chem. Rev., 2013, 113, 119–191 CrossRef PubMed.
- Y. V. Il'ichev and J. Wirz, J. Phys. Chem. A, 2000, 104, 7856–7870 CrossRef.
- B. L. Frey, Y. Lin, M. S. Westphall and L. M. Smith, J. Am. Soc. Mass Spectrom., 2005, 16, 1876–1887 CrossRef CAS PubMed.
- https://omlc.org/index.html .
- https://webbook.nist.gov .
- J. Laure, R. Antoine, A.-R. Allouche, M. Broyer, J. Lemoine and D. Philippe, J. Am. Chem. Soc., 2007, 129, 8428–8429 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9cc05712a |
| This journal is © The Royal Society of Chemistry 2019 |