
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A light-responsive liposomal agent for MRI contrast enhancement and monitoring of cargo delivery†
F.
Reeßing
ab,
M. C. A.
Stuart
 b,
D. F.
Samplonius
c,
R. A. J. O.
Dierckx
a,
B. L.
Feringa
ab,
W.
Helfrich
c and
W.
Szymanski
*ab
b,
D. F.
Samplonius
c,
R. A. J. O.
Dierckx
a,
B. L.
Feringa
ab,
W.
Helfrich
c and
W.
Szymanski
*ab
aDepartment of Radiology, Medical Imaging Center, University of Groningen, University Medical Center Groningen, Hanzeplein 1, 9713GZ Groningen, The Netherlands. E-mail: w.szymanski@umcg.nl
bStratingh Institute for Chemistry, University of Groningen, Nijenborgh 4, 9747 AG Groningen, The Netherlands
cTranslational Surgical Oncology, Department of Surgery, University of Groningen, University Medical Center Groningen, Hanzeplein 1/BA44, 9713GZ Groningen, The Netherlands
First published on 21st August 2019
Abstract
Medical magnetic resonance imaging (MRI) produces high-resolution anatomical images of the human body, but has limited capacity to provide useful molecular information. The light-responsive, liposomal MRI contrast agent described herein could be used to provide an intrinsic theranostic aspect to MRI and enable tracking the distribution and cargo release of drug delivery systems upon light-triggered activation.
In the clinic, Magnetic Resonance Imaging (MRI) is widely used as a non-invasive medical imaging technique that provides anatomical information with excellent resolution, without exposing the patient to ionizing radiation.1 The contrast in MRI stems from the difference in local densities and relaxation times of protons in tissues. In ∼30 million clinical scans performed annually worldwide, the contrast is further enhanced by the administration of paramagnetic contrast agents (CAs), such as gadolinium(III) complexes, which significantly shorten the T1 relaxation time of surrounding protons.2–4 This causes a higher intensity in the T1-weighted MR image and enables the visualization of the distribution of the CA in the human body.
Tissue-specific CAs are currently available to image structures that are barely distinguishable on a regular scan, such as the vascularization of the brain.5 However, due to the low sensitivity of MRI, the requirement of relatively high (>0.01 mM) local concentrations of CAs for effective signal enhancement presents a major limitation, especially regarding the development of CAs for the imaging of disease-specific biomarkers that are present at much lower concentrations. Therefore – while structures that are highly abundant in the human body, such as fibrin or collagen, can be readily visualized6–10 – targeted imaging of less abundant receptors or other proteins that are associated with certain pathological conditions, remains challenging.
This problem has been previously addressed through the development of responsive CAs, that show increased contrast enhancement upon activation by enzymes, ions, neurotransmitters or that take advantage of changes in e.g. pH, temperature or redox potential.4,10–16 Even though the effectiveness of this strategy has been proven for these targets, certain limitations to this approach remain: for instance, the untimely and/or off-target activation, as the conditions for the activation of the responsive CAs are frequently also present outside the lesion(s) in normal, healthy tissues.
In this respect, local activation of a CA with light could be used as a general strategy for improved MRI contrast enhancement. Of note, the use of photons as CA activators would not interfere with endogenous physiological processes.17 Moreover, light can be delivered with high spatiotemporal resolution and is biocompatible within a broad wavelength range.18,19 Due to these advantages, the research fields focusing on the use of light for biomedical applications, e.g. photopharmacology,20 photodynamic therapy (PDT),21 or optogenetics,22 are expanding very quickly fueled by promising results. As compared to other stimuli, such as ultrasound or heat, the use of light also presents specific challenges, including the limited penetration depth and potential toxicity.
The research presented here aims to establish a general strategy for signal amplification in contrast-enhanced MRI, which could be used for selective imaging of low-concentration targets. This strategy envisions the use of targeted light-emitting systems that locally activate the MRI CA, resulting in signal amplification. A key advantage of this approach is that the use of light for activation provides a CA that is readily adaptable to various targets by changing the light-emitting system, in contrast to the systems that are limited to one specific target.
As a key step towards this general goal, we describe here the synthesis and evaluation of a photoactivated MRI CA that changes its relaxivity in response to irradiation with blue light. Furthermore, we show how this liposomal CA can simultaneously be used as a responsive cargo delivery system (Fig. 1).23
 | ||
| Fig. 1 Design principle for light-activated MRI contrast agents for imaging (A) and theranostics (B). The Gd(III)-complex for T1-signal enhancement is incorporated into the bilayer of liposomes. Upon irradiation with λ = 400 nm light, the complex is released, causing a decrease in T1 relaxivity. (A) A targeting moiety (here an antibody) binding to the target tissue bears a light-emitting system that leads to the release of the gadolinium complex from the lipid bilayer of the liposomes. (B) The liposomal CA can be used for site-selective drug delivery using local irradiation as a stimulus to release the liposome cargo. Upon light irradiation, the liposomes concurrently release the Gd(III) complex and the payload incorporated in their aqueous lumen. | ||
For the successful design of a photoactivatable CA, it is crucial to consider the molecular characteristics influencing its relaxivity,24 such as (i) tumbling time, and (ii) number and (iii) residence time of water molecules coordinated to the gadolinium complex.8 Control over of the first two features is straightforward and was used for the design of responsive CAs.25 In line with the enzyme-based approach by Aime and co-workers,26 we designed a CA that, upon light-activation, converts from a relatively large nanoscopic complex to a small molecule, resulting in a significant change in relaxivity.
The design relies on linking a gadolinium complex, via a photocleavable group, to a lipophilic alkyl chain, which functions as an anchoring group for liposomes (Fig. 2). We hypothesized that irradiation of such liposomes would induce photocleavage and subsequent release of the gadolinium complex with an additional free carboxylic acid group (Fig. 2). This process leads to a lengthening of the T1 relaxation time. The reasons behind the signal change include the modulation of the tumbling time, as well as a change in the hydration state, since the liberated carboxylate moiety may coordinate to the gadolinium ion replacing one water molecule from the complex. Since it is generally preferred to obtain an increase in signal upon activation, we envision a ratiometric approach, analyzing the T1 and T2 relaxation time, for future applications, following the example of Aime et al.27
 | ||
| Fig. 2 Molecular structure of the gadolinium complex of compound 1 (Gd-1) and its photo-product Gd-2. | ||
To achieve an efficient, short and high-yielding synthesis, we used a Passerini multicomponent reaction (MCR) for creating the photoactive scaffold28 that could in subsequent transformations be modified with a liposome-anchoring group and a chelator for gadolinium yielding compound 1 (Fig. 3).
 | ||
| Fig. 3 Summary of key synthetic steps in the synthesis of compound 1: Passerini MCR for the synthesis of the photoactive scaffold, an azide–alkyne cycloaddition for attachment of the alkyl chains and a nucleophilic substitution for introduction of the gadolinium ligand. | ||
The liposomes were prepared with an equimolar mixture of compound 1 and 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC) in TBS buffer (pH 7.5). By adding Gd(III) to the pre-formed liposomes, we assume to form the complex with the ligands facing to the outside only, resulting in the photo-triggered release of the Gd(III) complex solely to outside and not the lumen. After removal of unselectively bound Gd(III) ions by dialysis, cryoTEM (Fig. S1, ESI†), dynamic light scattering analysis (Fig. S7, ESI†) and EDX spectroscopy (Fig. S1, ESI†) confirmed the formation of small unilamellar vesicles and accumulation of gadolinium in them. The concentration of gadolinium in the sample, determined by ICP-OES, was 0.95 mM, indicating that the complex was formed with 76% of all available ligands, assuming that the complex was only formed with the ligands facing outside.
We further used the FFC NMR relaxometry to confirm the synthetic reproducibility, stability over up to 4 weeks and photoresponsiveness of the liposome formulation (Fig. 4B, C and Fig. S2, ESI†). Next, we examined the effect of exposure to light (λ = 400) nm on the relaxation rate. Irradiation results in a marked decrease in relaxivity within 1 h of irradiation (Fig. 4A). Already after 10 min, a change ΔT1 of 21% (measured at 10 MHz) was observed, which is comparable to values reported for other light-switchable paramagnetic metal complexes.29–31 Moreover, the decrease in relaxivity coincided with a change in the shape of the NMRD profile from the one characteristic for macromolecular or nanoscopic contrast agents with an increase at higher field strength (>7 MHz) to the one of a small molecule CA,26 thus indicating the successful uncaging of the gadolinium complex from the liposome. With prolonged irradiation (for 60 min in total), the decrease in relaxivity could be further enhanced to 49% of the initial value (from 10.7 mM−1 s−1 to 5.2 mM−1 s−1). Likewise, the relaxation rate at 4.7 T, which is closer to the operating field of (pre-)clinical MRI scanners, decreased by 61% after 60 min irradiation (Fig. S4, ESI†).
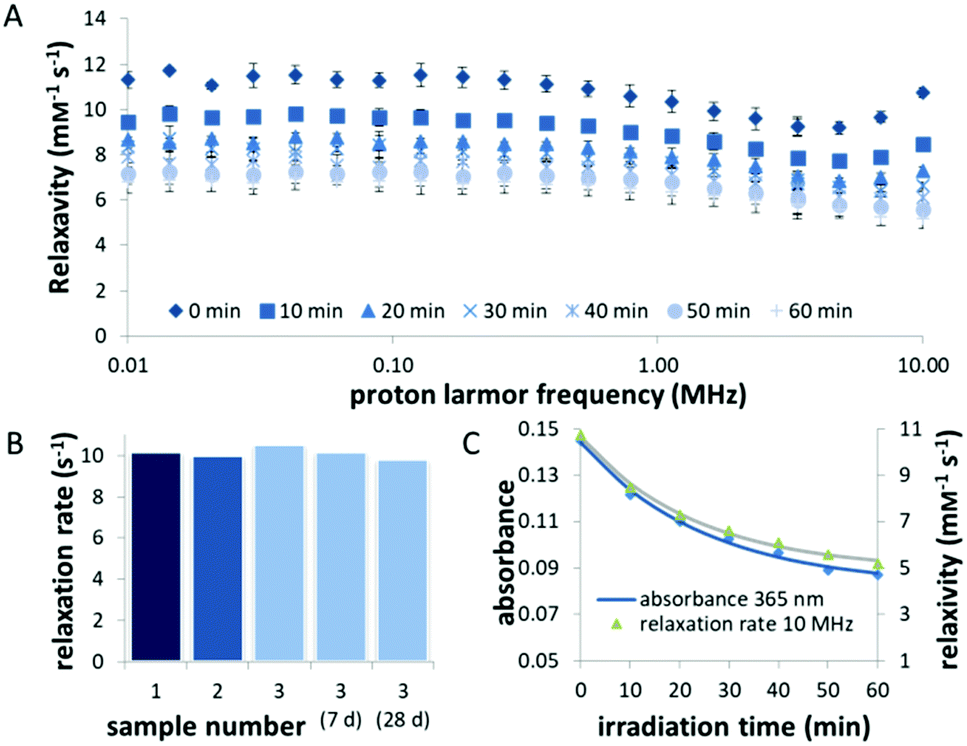 | ||
| Fig. 4 Stability and photochemical analysis of the liposomes containing Gd-1. (A) Average NMRD profile curves of samples 1–3 before and after irradiation with light (λ = 400 nm) for indicated times. The results show the average of three measurements of independently prepared samples. (B) The relaxation rates measured at 10 MHz of three independently prepared samples (1–3). The relaxation rate of sample 3 did not change substantially for up to 28 days of storage. (C) Average decrease in relaxivity recorded at 10 MHz compared to decrease in absorbance at λ = 365 nm. | ||
Correlation of the kinetics of the relaxivity decrease, measured by FFC relaxometry, and the uncaging process, followed by UV-vis spectroscopy, affirms that the change in relaxivity stems from the photocleavage of compound Gd-1 docked into the liposomal bilayer (Fig. 4C).
A major concern of the application of Gd-based CAs is the instability of the Gd(III) complex, as free Gd(III) has long-term toxic effects on the human body.3 While cyclic complexes are generally considered to be stable,4,32,33 we nevertheless investigated the stability of our Gd(III) complex upon irradiation, employing a photometric assay in which xylenol orange is used as a sensitive indicator for the presence of free Gd(III).34 We found no substantial increase in free Gd(III) concentration after 1 h of irradiation with blue light (Fig. S5, ESI†).
To further validate the applicability of the presented CA in a biological setting, we evaluated the toxicity of the liposome formulation and its photo-products towards human umbilical vein endothelial cells (HUVEC), human normal epithelial cells and M1 macrophages. No cytotoxic effect of liposomes that contained Gd-1 in their bilayer and that were either kept in the dark or pre-irradiated for up to 60 min, as compared to medium control, were detected on the cell lines (Fig. S8, ESI†).
Next, we explored the possibility of using the liposomal CA for MRI-guided drug delivery. To this end, we evaluated whether cleavage of compound 1 destablizes the lipid bilayer and thereby promotes the release of the liposome cargo. Calcein, a fluorescent dye, was encapsulated into the aqueous lumen of liposomes decorated with 1 at high, self-quenching concentrations (0.1 M). Only upon irradiation with λ = 400 nm light, a clear increase in fluorescence was observed (Fig. 5), due to the release and dilution of calcein35–37 indicating the destabilization of the bilayer. Unfortunately, it was not possible to determine the exact release rate due to calcein photobleaching.38 DLS showed re-organization of the liposomes, leading to a net decrease in size (Fig. S7, ESI†).
 | ||
| Fig. 5 Evaluation of the effect of photocleavage on liposome integrity. Fluorescence intensity of 50% DOPC/50% compound 1 liposomes, loaded with calcein at self-quenching concentration (0.1 M), measured as a technical triplicate. Upon irradiation, membrane integrity is reduced as is evident from an increase in fluorescence due to calcein release. | ||
We present here the proof of principle for an activated MRI CA with intrinsic capability for drug delivery, offering prospects for diagnostics and image-guided therapy. We developed and evaluated a light-responsive liposomal gadolinium complex and we demonstrated that its exposure to light results in a marked decrease in relaxation rate, indicating the conversion of a nanoscopic object into a small molecule.
The increase in the permeability of the liposomes upon light exposure opens new possibilities to employ this CA for theranostic applications. To date, there are only few examples of agents combining MR-imaging with pharmacotherapy,23,39 including thermo-sensitive release of MRI CA and therapeutics from liposomes and a combination of Gd(III) complexes with porphyrins for PDT.40,41 Our strategy, however, stands out due to the prospect of using internal light-emitting targeting moieties for activation, which makes the drug release system unbiased and independent of external stimuli.
In further perspective, we envision to use a two-step approach in which the patient is first injected with a disease-specific antibody (or derivate thereof) equipped with a bioluminescent enzyme–substrate system. After its injection, the conjugate is allowed to selectively accumulate in the lesion(s). Subsequently, the corresponding substrate of the light-producing enzyme is injected and converted at the site of the lesion only, resulting in the localized generation of photons. In turn, these photons locally enhance MRI contrast of the light-activatable CA. Deliberate timing of the scans and stepwise administration of the respective components would result in distinguished resolution. Possible luminescent tools, that could be used for the activation of the CA, are luciferin/luciferase systems, which have been optimized for in vivo bioluminescence imaging42,43 or horseradish peroxidase, which has been expressed in mammalian cells and explored for local prodrug activation in vivo.44 Altogether, this method may be useful to reduce the side effects in systemic chemotherapy for the treatment of localized malignant disease.
For the clinical development of the CA reported here, it is crucial to shift the activation wavelength to >600 nm, to maximize tissue penetration and reduce light-associated toxicity.18 Recent developments in green and red light-responsive photocaging groups offer efficient activation within a clinical setting.45–48 With regards to the use of our system for MRI guided drug delivery, a bathochromic shift in activation wavelength would open up the possibility to use clinically established light delivery systems,49–51 commonly used in PDT or photothermal therapies, for triggering the drug release.
The financial support from the Dutch Organization for Scientific Research (VIDI grant no. 723.014.001 for W. S.), the Dutch Cancer Society (grant RUG2014-6986 for W. H.) and the Dutch Ministry of Education, Culture and Science (Gravitation program 024.001.035 for B. L. F.) is gratefully acknowledged. We thank Ms Verena Böhmer for helpful discussions, eng. Theodora D. Tiemersma-Wegman for MS analysis, eng. Hans van der Velde for ICP-OES analysis, Pieter van der Meulen for help with determination of relaxivity at 4.7 T and Mark A. J. M. Hendriks for the macrophages cytotoxicity assay.
Conflicts of interest
The authors declare no conflict of interests.Notes and references
- D. Hao, T. Ai, F. Goerner, X. Hu, V. M. Runge and M. Tweedle, J. Magn. Reson. Imaging, 2012, 36, 1060–1071 CrossRef PubMed.
- J. Lohrke, T. Frenzel, J. Endrikat, F. C. Alves, T. M. Grist, M. Law, J. M. Lee, T. Leiner, K.-C. Li, K. Nikolaou, M. R. Prince, H. H. Schild, J. C. Weinreb, K. Yoshikawa and H. Pietsch, Adv. Ther., 2016, 33, 1–28 CrossRef PubMed.
- J. Garcia, S. Z. Liu and A. Y. Louie, Philos. Trans. R. Soc., A, 2017, 375, 20170180 CrossRef PubMed.
- J. Wahsner, E. M. Gale, A. Rodríguez-Rodríguez and P. Caravan, Chem. Rev., 2019, 119, 957–1057 CrossRef CAS PubMed.
- E. Boros, E. M. Gale and P. Caravan, Dalton Trans., 2015, 44, 4804–4818 RSC.
- F. OukhatarukhatMeudal, C. Landon, N. K. Logothetis, C. Platas-Iglesias, G. Angelovski and É. Tóth, Chem. – Eur. J., 2015, 21, 11226–11237 CrossRef PubMed.
- K. Overoye-Chan, S. Koerner, R. J. Looby, A. F. Kolodziej, S. G. Zech, Q. Deng, J. M. Chasse, T. J. McMurry and P. Caravan, J. Am. Chem. Soc., 2008, 130, 6025–6039 CrossRef CAS PubMed.
- L. Helm, A. E. Merbach and E. Tóth, The chemistry of contrast agents in medical magnetic resonance imaging, Wiley, 2nd edn, 2013 Search PubMed.
- P. A. Waghorn, C. M. Jones, N. J. Rotile, S. K. Koerner, D. S. Ferreira, H. H. Chen, C. K. Probst, A. M. Tager and P. Caravan, Angew. Chem., Int. Ed., 2017, 56, 9825–9828 CrossRef CAS.
- D. V. Hingorani, A. S. Bernstein and M. D. Pagel, Contrast Media Mol. Imaging, 2015, 10, 245–265 CrossRef CAS.
- F. A. Rojas-Quijano, G. Tircsó, E. Tircsóné Benyó, Z. Baranyai, H. Tran Hoang, F. K. Kálmán, P. K. Gulaka, V. D. Kodibagkar, S. Aime, Z. Kovács and A. D. Sherry, Chem. – Eur. J., 2012, 18, 9669–9676 CrossRef CAS PubMed.
- M. Lepage, W. C. Dow, M. Melchior, Y. You, B. Fingleton, C. C. Quarles, C. Pépin, J. C. Gore, L. M. Matrisian and J. O. McIntyre, Mol. Imaging, 2007, 6, 393–403 CrossRef CAS PubMed.
- A. Louie, J. Magn. Reson. Imaging, 2013, 38, 530–539 CrossRef PubMed.
- J. Lux and A. D. Sherry, Curr. Opin. Chem. Biol., 2018, 45, 121–130 CrossRef CAS PubMed.
- K. D. Verma, J. O. Massing, S. G. Kamper, C. E. Carney, K. W. MacRenaris, J. P. Basilion and T. J. Meade, Chem. Sci., 2017, 8, 5764–5768 RSC.
- K. W. MacRenaris, Z. Ma, R. L. Krueger, C. E. Carney and T. J. Meade, Bioconjugate Chem., 2016, 27, 465–473 CrossRef CAS PubMed.
- Y. Tang, X. Lu, C. Yin, H. Zhao, W. Hu, X. Hu, Y. Li, Z. Yang, F. Lu, Q. Fan and W. Huang, Chem. Sci., 2019, 10, 1401–1409 RSC.
- R. Weissleder and V. Ntziachristos, Nat. Med., 2003, 9, 123–128 CrossRef CAS PubMed.
- W. A. Velema, W. Szymanski and B. L. Feringa, J. Am. Chem. Soc., 2014, 136, 2178–2191 CrossRef CAS PubMed.
- F. Reeßing and W. Szymanski, Curr. Med. Chem., 2018, 24, 4905–4950 CrossRef PubMed.
- C. A. Robertson, D. H. Evans and H. Abrahamse, J. Photochem. Photobiol., B, 2009, 96, 1–8 CrossRef CAS PubMed.
- L. Fenno, O. Yizhar and K. Deisseroth, Annu. Rev. Neurosci., 2011, 34, 389–412 CrossRef CAS PubMed.
- F. Reeβing and W. Szymanski, Curr. Opin. Biotechnol., 2019, 58, 9–18 CrossRef PubMed.
- P. Caravan, J. J. Ellison, T. J. McMurry and R. B. Lauffer, Chem. Rev., 1999, 99, 2293–2352 CrossRef CAS PubMed.
- C. S. Bonnet and É. Tóth, Chim. Int. J. Chem., 2016, 70, 102–108 CrossRef CAS PubMed.
- V. Catanzaro, C. V. Gringeri, V. Menchise, S. Padovan, C. Boffa, W. Dastrù, L. Chaabane, G. Digilio and S. Aime, Angew. Chem., Int. Ed., 2013, 52, 3926–3930 CrossRef CAS PubMed.
- S. Aime, F. Fedeli, A. Sanino and E. Terreno, J. Am. Chem. Soc., 2006, 128, 11326–11327 CrossRef CAS PubMed.
- W. Szymański, W. A. Velema and B. L. Feringa, Angew. Chem., Int. Ed., 2014, 53, 8682–8686 CrossRef PubMed.
- C. Tu, E. A. Osborne and A. Y. Louie, Tetrahedron, 2009, 65, 1241–1246 CrossRef CAS PubMed.
- C. Tu and A. Y. Louie, Chem. Commun., 2007, 1331 RSC.
- G. Heitmann, C. Schütt, J. Gröbner, L. Huber and R. Herges, Dalton Trans., 2016, 45, 11407–11412 RSC.
- P. Hermann, J. Kotek, V. Kubíček and I. Lukeš, Dalton Trans., 2008, 3027 RSC.
- T. Kanda, M. Osawa, H. Oba, K. Toyoda, J. Kotoku, T. Haruyama, K. Takeshita and S. Furui, Radiology, 2015, 275, 803–809 CrossRef.
- A. Barge, G. Cravotto, E. Gianolio and F. Fedeli, Contrast Media Mol. Imaging, 2006, 1, 184–188 CrossRef.
- J. N. Weinstein, S. Yoshikami, P. Henkart, R. Blumenthal and W. A. Hagins, Science, 1977, 195, 489–492 CrossRef CAS PubMed.
- T. Shimanouchi, P. Walde, J. Gardiner, Y. R. Mahajan, D. Seebach, A. Thomae, S. D. Krämer, M. Voser and R. Kuboi, Biochim. Biophys. Acta, Biomembr., 2007, 1768, 2726–2736 CrossRef CAS PubMed.
- W. Deng, W. Chen, S. Clement, A. Guller, Z. Zhao, A. Engel and E. M. Goldys, Nat. Commun., 2018, 9, 2713 CrossRef PubMed.
- K. E. Roberts, A. K. O’Keeffe, C. J. Lloyd and D. J. Clarke, J. Fluoresc., 2003, 13, 513–517 CrossRef CAS.
- S. Lacerda and É. Tóth, ChemMedChem, 2017, 12, 883–894 CrossRef CAS PubMed.
- A. Sour, S. Jenni, A. Ortí-Suárez, J. Schmitt, V. Heitz, F. Bolze, P. Loureiro de Sousa, C. Po, C. S. Bonnet, A. Pallier, É. Tóth and B. Ventura, Inorg. Chem., 2016, 55, 4545–4554 CrossRef CAS PubMed.
- M. de Smet, S. Langereis, S. van den Bosch and H. Grüll, J. Controlled Release, 2010, 143, 120–127 CrossRef CAS PubMed.
- T. Xu, D. Close, W. Handagama, E. Marr, G. Sayler and S. Ripp, Front. Oncol., 2016, 6, 150 Search PubMed.
- D. M. Close, S. S. Patterson, S. Ripp, S. J. Baek, J. Sanseverino and G. S. Sayler, PLoS One, 2010, 5, e12441 CrossRef PubMed.
- J. Tupper, M. R. Stratford, S. Hill, G. M. Tozer and G. U. Dachs, Cancer Gene Ther., 2010, 17, 420–428 CrossRef CAS PubMed.
- K. Sitkowska, B. L. Feringa and W. Szymański, J. Org. Chem., 2018, 83, 1819–1827 CrossRef CAS PubMed.
- T. Slanina, P. Shrestha, E. Palao, D. Kand, J. A. Peterson, A. S. Dutton, N. Rubinstein, R. Weinstain, A. H. Winter and P. Klán, J. Am. Chem. Soc., 2017, 139, 15168–15175 CrossRef CAS PubMed.
- X. Wang and J. A. Kalow, Org. Lett., 2018, 20, 1716–1719 CrossRef CAS PubMed.
- N. Rubinstein, P. Liu, E. W. Miller and R. Weinstain, Chem. Commun., 2015, 51, 6369–6372 RSC.
- J. M. Silva, E. Silva and R. L. Reis, J. Controlled Release, 2019, 298, 154–176 CrossRef CAS.
- L. Brancaleon and H. Moseley, Lasers Med. Sci., 2002, 17, 173–186 CrossRef CAS.
- M. A. Calin and S. V. Parasca, Lasers Med. Sci., 2009, 24, 453–460 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9cc05516a |
| This journal is © The Royal Society of Chemistry 2019 |