
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Optimised oligonucleotide substrates to assay XPF-ERCC1 nuclease activity for the discovery of DNA repair inhibitors†
Adam M.
Thomas‡
 abc,
Sanja
Brolih‡
b,
Joanna F.
McGouran‡§
a,
Afaf H.
El-Sagheer
ad,
Denis
Ptchelkine
b,
Morgan
Jones
c,
Neil Q.
McDonald
*ce,
Peter J.
McHugh
*b and
Tom
Brown
*a
abc,
Sanja
Brolih‡
b,
Joanna F.
McGouran‡§
a,
Afaf H.
El-Sagheer
ad,
Denis
Ptchelkine
b,
Morgan
Jones
c,
Neil Q.
McDonald
*ce,
Peter J.
McHugh
*b and
Tom
Brown
*a
aDepartment of Chemistry, University of Oxford, Chemistry Research Laboratory, 12 Mansfield Road, Oxford, OX1 3TA, UK. E-mail: tom.brown@chem.ox.ac.uk; Tel: +44 01865 275413
bDepartment of Oncology, MRC-Weatherall Institute of Molecular Medicine, University of Oxford, John Radcliffe Hospital, Oxford, OX3 9DS, UK
cThe Francis Crick Institute, 1 Midland Road, London NW1 1AT, UK
dChemistry Branch, Department of Science and Mathematics, Faculty of Petroleum and Mining Engineering, Suez University, Suez 43721, Egypt
eInstitute of Structural and Molecular Biology, Department of Biological Sciences, Birkbeck College, Malet Street, London WC1E 7HX, UK
First published on 9th September 2019
Abstract
We report the design and optimisation of novel oligonucleotide substrates for a sensitive fluorescence assay for high-throughput screening and functional studies of the DNA repair enzyme, XPF-ERCC1, with a view to accelerating inhibitor and drug discovery.
Many chemotherapy drugs kill tumour cells through the induction of DNA damage; DNA interstrand crosslinks (ICLs) are particularly toxic to cells as they prevent DNA replication and transcription.1 DNA cross-linking drugs, including platinum-based drugs, are used to treat a range of tumours including many hard-to-treat cancers (such as pancreatic, oesophageal and lung) for which survival rates remain low.1,2 DNA repair pathways directly combat ICL-inducing therapies by removing the toxic DNA damage they induce,2–5 where resistance to therapy remains a major clinical problem, limiting the effective use of chemotherapeutic drugs.6
Targeting DNA repair is one of the most compelling, and poorly exploited strategies for sensitising tumours to ICL-inducing agents and other chemotherapies.7–9 In particular, increases in levels of ERCC1 have been linked to poor response to chemotherapy across several cancer types in multiple clinical studies.10–13 XPF exists as an obligate heterodimer with ERCC1, forming a structure selective endonuclease, where XPF is the catalytic subunit, which preferentially cleaves DNA duplexes adjacent to a 3′ single-stranded flap.14–17 XPF-ERCC1 is involved in a number of DNA repair pathways including nucleotide excision repair (NER), homologous recombination (HR) and ICL repair, playing a critical role in the repair-response to a number of DNA damaging agents used in cancer therapy.18
Despite the urgent need to explore inhibition of XPF-ERCC1 as a therapeutic avenue, few compounds have been identified for this purpose.19,20 This is in part due to limited access to systematically optimised substrates, necessary for highly robust and sensitive screening assays. Moreover, several screens for XPF-ERCC1 inhibitors have also been limited to screening in silico or use truncated protein forms, leading to low hit numbers and incomplete characterisation.20,21
The aim of this work was to generate and validate a highly sensitive, scalable, robust assay for XPF-ERCC1 activity with a broad dynamic range for use in high-throughput inhibitor screens. The assay we describe utilises full-length XPF-ERCC1 and fork DNA structures which give high DNA duplex stability whilst conferring sensitivity. The position of the fluorophore and quencher relative to the junction has been varied to optimise this assay, and importantly the use of full-length XPF-ERCC1 gives potential for optimal inhibitor selectivity in comparison to truncated protein, as well as the opportunity to explore other modes of inhibition, such as allosteric approaches. Full-length XPF-ERCC1 complex was purified from insect cells using a baculovirus expression system [Fig. S1A (ESI†)].22 An XPF active site substitution mutant D676A (XPFD676A-ERCC1), devoid of nuclease activity, was also purified as a control for contaminating insect cell nuclease activity.
We initially characterised the activity of XPF-ERCC1 using a ‘simple fork’ structure. This was labelled at the 3′-terminus of the single-stranded arm with a fluorescent CY3, a bright and stable fluorophore (Fig. 1A). The assays employed a fixed concentration of the divalent cations previously reported to support the activity of XPF-ERCC1, (Mg2+ and Mn2+) and increasing concentrations of XPF-ERCC1 within the range previously reported to be active on this type of substrate (10 to 70 nM), with a 60 minute incubation using a fixed, excess concentration of substrate (100 nM) (Fig. 1B). Activity could be observed for XPF-ERCC1 at concentrations as low as 10 nM with greater activity in the Mn2+ buffer. As expected, XPFD676A-ERCC1 (lanes marked: ND) supported no activity in either buffer when present at the highest concentration (70 nM). There have been reports of analysis of XPF-ERCC1 activities at both 30 °C and 37 °C. Here, we determined the fork substrate to be equivalently digested by 40 nM XPF-ERCC1 at both temperatures (Fig. S1B, ESI†), suggesting that activity is independent of temperature within this range.
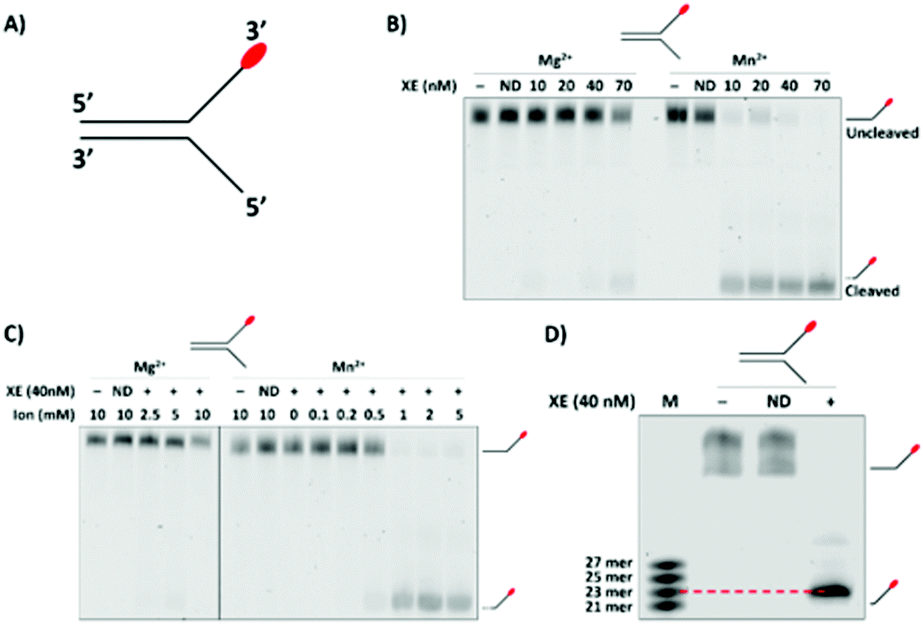 | ||
| Fig. 1 (A) Control fork. The 3′- and 5′-direction are consistent throughout. (B) Validation of XPF-ERCC1 (XE) activity on a known substrate at varying XE concentrations and in the presence of 2 mM of Mn2+ or Mg2+. Incubation was at 37 °C for 60 min in the presence of the control fork (100 nM). The XPFD676A-ERCC1 inactive ‘nuclease dead’ (ND) form of XE was used as a control to demonstrate that nuclease activity is intrinsic to the XE protein. (C) Determination of optimum divalent cation species and concentration. The control fork (100 nM) was incubated with XE (40 nM), and varying concentrations of Mg2+ or Mn2+, for 60 min at 37 °C. For both (B) and (C) the samples were analysed on a 10% denaturing PAGE gel. (D) High-resolution 20% denaturing gel where substrate and reaction product were run alongside multiple marker oligonucleotides with molecular weights close to the predicted XE product size to determine the major cleavage site on the control fork. Reactions were performed as in panel (A) and (B), with 1 mM Mn2+. ND = XPFD676A-ERCC1. | ||
The data presented in Fig. 1B implied that a major determinant of XPF-ERCC1 activity within our system is the divalent cation present. We consequently explored XPF-ERCC1 activity over a range of both Mg2+ and Mn2+ concentrations. XPF-ERCC1 activity is maximal at 1 mM Mn2+, while only weak activity is observed at concentrations up to 10 mM Mg2+ (Fig. 1C and Fig. S1C, ESI†), as previously rereported.16 We therefore employed Mn2+, which supports greater XPF nuclease activity, at 1 mM for further optimisation experiments. Finally, we carefully mapped the major cleavage site of XPF-ERCC1 on this simple 3′-fluor-labelled fork substrate by generating a marker ladder containing 3′-fluor-labelled oligonucleotides of 2-nucleotide incremental sizes that flank the predicted incision position. This revealed that the majority (over 90%) of released products were 23-nucleotides in length, corresponding to a cleavage site 2-nucleotides 5′ to the fork junction (Fig. 1D).
Having established optimal conditions for XPF-ERCC1 analysis, we varied the position of the fluorophore within the single-stranded arm of our fork substrates and analysed the effect on activity. This was in anticipation of including a quencher moiety on the strand opposing the fluorescently labelled strand. This would permit the establishment of a real-time fluorescence incision assay, in which dsDNA cleaving by XPF-ERCC1 uncouples the fluorophore and quencher leading to increased fluorescence. This readout can subsequently be used to quantify enzyme activity and derive kinetic measurements.
In order that the fluorescent moiety can be uncoupled from the quencher incorporated elsewhere in the substrate, it is necessary to place the fluorophore on one of the substrate arms, distal to the major XPF-ERCC1 cut site, mindful that the location of the fluorophore might itself further qualitatively and quantitatively modulate the activity of XPF-ERCC1. Moreover, in order to optimally position both a fluorophore and quencher on the substrates, as became apparent (below), it might be necessary to move the fluorophore from the extreme 3′-terminus of the substrate arms. Consequently, we generated a panel of substrates with a fluorophore positioned 8-nucleotides, 4-nucleotides and 2-nucleotides 3′ to the fork junction, and determined the activity of XPF-ERCC1 on these with reference to the previously optimised substrate harbouring a terminal 3′-fluorophore. All the substrates were efficiently digested by 10 nM XPF-ERCC1, although we detected a small shift in the product size for the substrate bearing the fluorophore two nucleotides from the fork junction (Fig. 2A). Further titration of XPF-ERCC1 revealed that full cleavage of each fork substrate occurred at concentrations between 10 and 20 nM in XPF-ERCC1 (Fig. S2, ESI†), and therefore 20 nM was selected as the lowest concentration for all further analysis. By running the reaction products on a gel adjacent to the molecular weight markers we confirmed that placing the moiety two nucleotides from the junction likely moves the incision position by a single nucleotide to a position 3-nucleotides from the junction (Fig. 2B). This implies that the presence of the fluorophore so close to the junction interferes sterically with the enzyme, altering its position of incision.
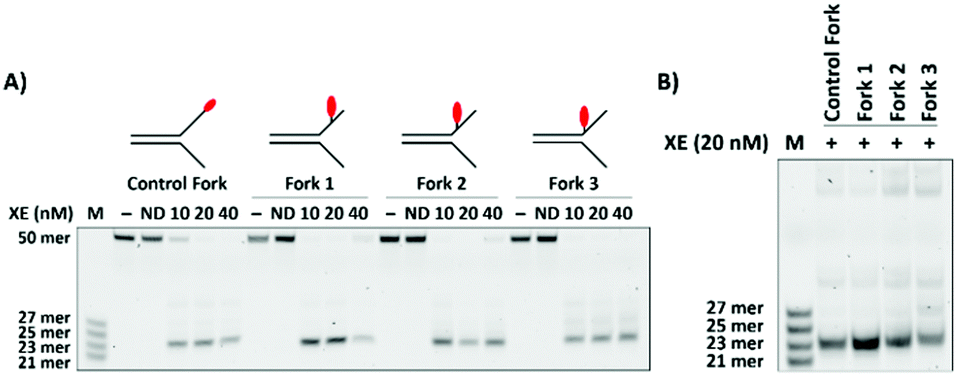 | ||
| Fig. 2 (A) Effect of varying the fluorophore position on the simple fork substrate. The fluorophore was positioned at 8, 4, and 2 nucleotides 3′ to the fork junction (Forks 1, 2 and 3, respectively) to evaluate whether it affected the activity of the XE. The different forks (100 nM) were incubated at 37 °C for 60 min, in the presence of 1 mM Mn2+ and varying concentrations of XE, and analysed on preheated 12.5% denaturing PAGE gels. (B) Direct comparison of major cleavage products of the four substrates is shown in panel A (Reactions performed and analysed as in panel A.) | ||
Having established that XPF-ERCC1 is equally capable of digesting substrates with the fluorophore at various positions along the fork arm, with the caveat that the incision site might be altered by positioning the fluorophore close to the junction, we next designed a panel of substrates containing a Black Hole Quencher (BHQ). This has excellent spectral overlap with the Quasar 570 dye on the opposing strand. The aim of this work was to generate a substrate that is digested with maximal efficiency by XPF-ERCC1 simultaneously providing a stable fluorescent signal associated with a low signal:noise and broad dynamic range. To achieve this we took an empirical approach, placing the BHQ quencher at various positions on the strand opposing the fluorophore, utilising substrates that contained a fluorophore 8-, 4-, and 2-nucleotides 3′ to the fork junction. Preliminary experiments suggested that placing the fluorophore at the 3′-terminus of the fork led to poor quenching for all BHQ positions due to the substantial spatial separation between the fluorophore and quencher when the latter is terminally located.
Examination of substrates containing the fluorophore 8-nucleotides 3′ to the fork junction combined with quencher located 5-nucleotides 3′ to the junction on the opposing strand (Fork 1.1), 3-nucleotides 3′ to the junction (Fork 1.2), at the junction (Fork 1.3) or 2-nucleotides 5′ to the junction (Fork 1.4) in the single strand region revealed an XPF-ERCC1 sensitivity to the quencher location (Fig. 3A). When the quencher is located within the duplex region, XPF-ERCC1 makes a series of incisions and loses selectivity, no longer producing a single major incision to release a 23-nucleotide product as observed with the control substrate lacking the quencher moiety. Nevertheless, despite the effect on cleavage position selectivity, the overall efficiency of substrate cleavage is not substantially impacted by the position of the quencher.
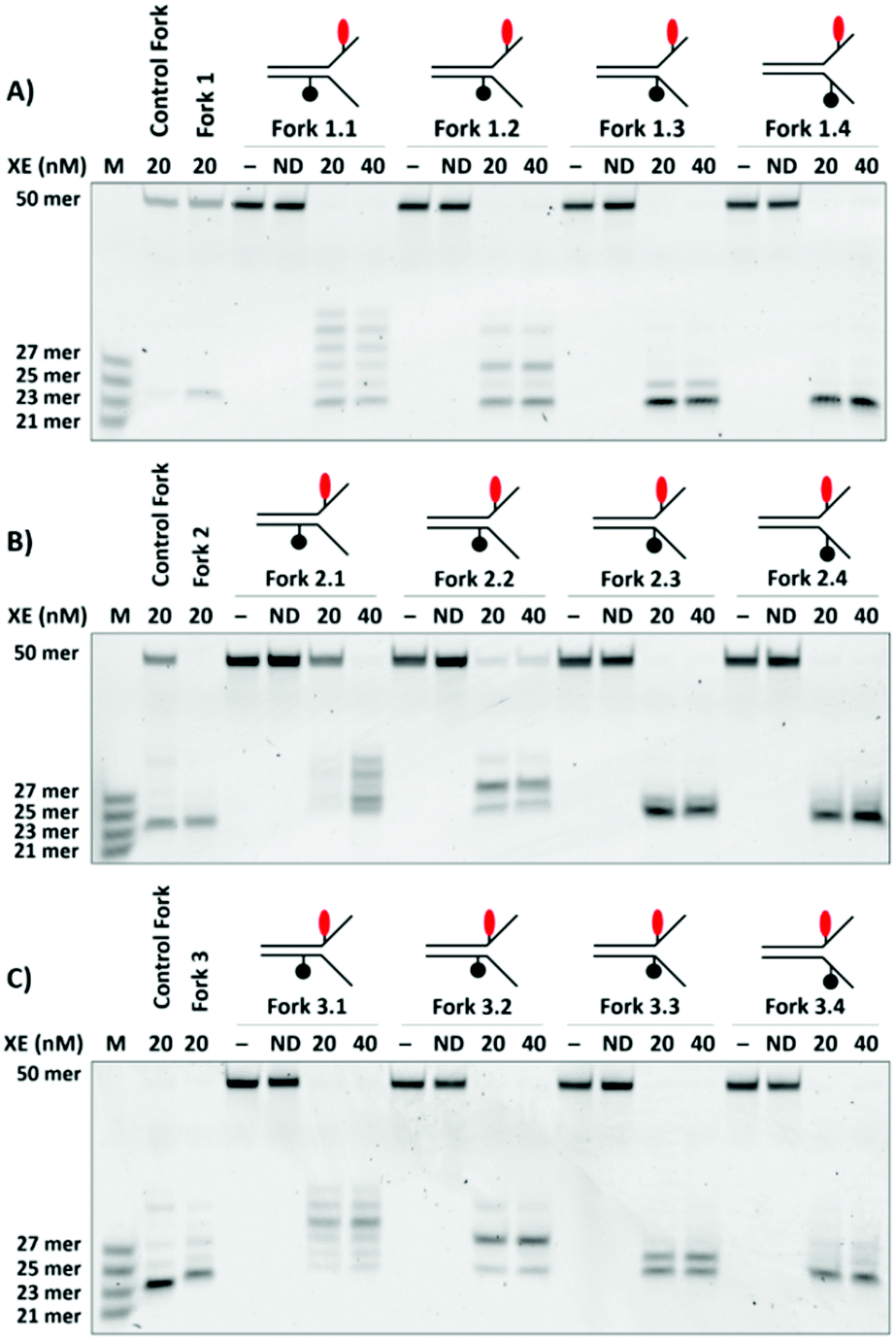 | ||
| Fig. 3 Effect of varying the Black Hole Quencher (BHQ) position on Forks 1, 2, and 3. Four positions, −5, −3, 0, and 2-nucleotides from the junction, on the strand opposing of that containing the fluorophore were functionalised with BHQ (Forks 1.1, 1.2, 1.3 and 1.4, respectively) and their effects on XE binding activity were evaluated. The assays were run and analysed under the conditions described in Fig. 2. The effect of varying BHQ location on Fork 1 is shown in panel A, on Fork 2 in panel B, where forks containing the quench −5, −3, 0 and 2-nucleotides from the junction are labelled Forks 2.1, 2.2, 2.3 and 2.4, respectively, and Fork 3 in panel C, where forks containing the quench −5, −3, 0 and 2-nucleotides from the junction are labelled Forks 3.1, 3.2, 3.3 and 3.4, respectively. | ||
Equivalent experiments were performed on substrates (Forks 2.1, 2.2, 2.3 and 2.4) containing the fluorophore four nucleotides to the 3′-side of the fork junction. This generated very similar data (Fig. 3B), where placement of the quencher within the duplex region produced a mixture of incision products. Finally, we examined the XPF-ERCC1 cleavage characteristics on substrates where the fluorophore was placed two nucleotides to the 3′-side of the fork junction in conjunction with the same set of quencher locations used in Fig. 3A and B (substrates Fork 3.1, 3.2, 3.3 and 3.4). These structures have the advantage that the fluorophore and quencher are in closer proximity than when the fluorophore is located closer to the 3′-terminus. However, a more complex pattern of incisions was observed for all substrates, regardless of the location of quencher (Fig. 3C). We propose that the presence of both a fluorophore and quencher in the vicinity of the fork junction region, a critical feature for substrate recognition and verification by XPF-ERCC1, interferes substantially with these processes, changing the cleavage site. A summary gel provides direct comparison of the qualitative and quantitative aspects of digestion characteristics (Fig. S3, ESI†). Based on the above analyses, we did not pursue any of the DNA structures where the quencher was located in the duplex region of the substrate (Forks 1.1, 1.2, 2.1, 2.2), as the multiple species could significantly reduce the robustness of the fluorogenic assay. In addition, none of the structures where the fluorophore is located 2-nucleotides from the junction, regardless of quencher combination, were further pursued since these also formed a complex array of products (3.1, 3.2, 3.3, 3.4).
High-throughput screens require non-gel-based assays. Development of a multi-well plate assay was necessary, where the generation of the fluorescent signal acts as a readout of XPF-ERCC1 activity. Initial assays using 100 nM DNA substrate showed reasonable signal:noise, and minimal fluorescence photobleaching. Optimisation led to the DNA concentration being raised to 200 nM, which was shown to increase the signal:noise, as well as the overall fluorescence readout. We then monitored the fluorogenic output of substrates 1.3, 1.4, 2.3 and 2.4 in a 384-well format as a function of time, with readings taken every minute for 30 min (Fig. 4A). Pleasingly all substrates produced little background fluorescence over this time course, indicating that all were stable under the reaction conditions, and that non-specific background fluorescence would not be a confounding feature in the assay. Substrates were further screened against varying concentrations of XPF-ERCC1, and this indicated that 40 nM was optimal for a robust high throughput assay (Fig. S4, ESI†).
 | ||
| Fig. 4 (A) Screening the selected fork substrates using a fluorescence plate reader to evaluate fluorogenic assay conditions and identify optimal fluorophore and quencher positioning. The selected forks (200 nM) were incubated at 37 °C for 30 min with either no XPF-ERCC1 (control) or XPF-ERCC1 (40 nM). These data were analysed using GraphPad Prism 7 (n = 3). (B) Signal-to-noise ratio of lead substrates. Assays were carried out as stated for panel 4A. Signal-to-noise ratio was calculated from 10 min, once fluorescence intensity had plateaued. The error bars indicate the standard error of mean (n = 3). (C) Readout stability of Fork 1.3. A completed 384 well assay (run for 30 min) was left for a further 135 min at room temperature and readings taken every 30 min The error bars indicate the standard error of mean (n = 3). (D) The broad, nuclease inhibitor N-hydroxy-1,8-naphthalimide (inset) was used to validate the assay. Fluorescence was determined on the 384 well platform over a range of inhibitor concentrations. Data were plotted and curve fitted using GraphPad Prism 7. The error bars indicate the standard error of mean (n = 3). | ||
Differing maximum fluorescence outputs were detected, where the substrate bearing the fluorescent moiety four nucleotides from the junction and the quencher at the junction (substrates 2.3) produced a lower signal than those where the quencher was positioned two nucleotides into the substrate arms (substrates 1.4 and 2.4), regardless of whether the fluorophore was located four or eight nucleotides from the junction. Fork 1.3 produced the best signal:noise (Fig. 4B) and the greatest dynamic range (Fig. 4A). It was therefore designated the preferred substrate for further validation.
A requirement for high-throughput screening is good temporal stability of the reaction substrate and products, as a substantial period of time often elapses between assay termination and plate reading during large-scale screens. We therefore measured the change in fluorescence of our optimal substrate (1.3) alone and following incubation with 10–40 nM XPF-ERCC1 for 60 min. After reaction termination by EDTA addition and incubation at room temperature for a further 135 minutes, the assay substrate and its digestion products were shown to be stable over the full extended period (Fig. 4C).
Finally, we validated our assay using a broad-spectrum small molecule nuclease inhibitor, N-hydroxy-1,8-naphthalimide (Fig. 4D). Employing the optimal substrate 1.3 in a 384 well assay format we determined XPF-ERCC1 activity as a function of inhibitor concentration. This produced an IC50 value of 32.9 μM (Fig. 4D), with a small standard error of the mean (1.1 μM) which is very close to the literature value of 30 μM.20,23
DNA repair nucleases are an untapped class of drug targets. Here we present a robust assay that has high signal:noise, broad dynamic range, high sensitivity and long-term stability. It will be useful for probing mechanistic enzymology and characterising the functional impact of mutations in XPF and interacting proteins such as SLX424 and XPA25 that are associated with the devastating inherited syndromes Xeroderma pigmentosum, Fanconi anaemia, Cockayne syndrome and Cerebro-Oculo-Facio-Skeletal syndrome.14
AT was supported by the GSK-Crick-Oxford Chemical Biology Doctoral Programme, EPSRC, BBSRC, and GSK. PJM and DP were supported by MRC Grant MR/R009368/1 and an MRC studentship to SB. N. Q. M. was supported by the Francis Crick Institute and Cancer Research UK (FC001115), the UK Medical Research Council (FC001115) and the Wellcome Trust (FC001115). AHES was supported by UK BBSRC grant BB/R008655/1.
Conflicts of interest
There are no conflicts to declare.Notes and references
- P. J. McHugh, V. J. Spanswick and J. A. Hartley, Lancet Oncol., 2001, 2, 483–490 CrossRef CAS PubMed.
- M. L. Dronkert and R. Kanaar, Mutat. Res., 2001, 486, 217–247 CAS.
- M. Räschle, P. Knipscheer, M. Enoiu, T. Angelov, J. Sun, J. D. Griffith, T. E. Ellenberger, O. D. Schärer and J. C. Walter, Cell, 2008, 134, 969–980 CrossRef PubMed.
- B. Sengerova, A. T. Wang and P. J. McHugh, Cell Cycle, 2011, 10, 3999–4008 CrossRef PubMed.
- J. Zhang and J. C. Walter, DNA Repair, 2014, 19, 135–142 CrossRef CAS PubMed.
- T. Helleday, E. Petermann, C. Lundin, B. Hodgson and R. A. Sharma, Nat. Rev. Cancer, 2008, 8, 193–204 CrossRef CAS.
- R. Plummer, Clin. Cancer Res., 2010, 16, 4527–4531 CrossRef CAS PubMed.
- M. J. O'Connor, Mol. Cell, 2015, 60, 547–560 CrossRef PubMed.
- L. Song, A. Ritchie, E. M. McNeil, W. Li and D. W. Melton, Pigm. Cell Melanoma Res., 2011, 24, 966–971 CrossRef CAS PubMed.
- K. Kirschner and D. W. Melton, Anticancer Res., 2010, 30, 3223–3232 CAS.
- Z. Zheng, T. Chen, X. Li, E. Haura, A. Sharma and G. Bepler, N. Engl. J. Med., 2007, 356, 800–808 CrossRef CAS PubMed.
- K. A. Olaussen, A. Dunant, P. Fouret, E. Brambilla, F. Andŕe, V. Haddad, E. Taranchon, M. Filiptis, R. Pirker, H. H. Popper, R. Stahel, L. Sabatier, J. P. Pignon, T. Tursz, T. Le Chevalier, J. C. Soria and IALT Bio Investigators, N. Engl. J. Med., 2006, 355, 983–991 CrossRef CAS PubMed.
- S. Postel-Vinay and J. C. Soria, J. Clin. Oncol., 2017, 35, 384–386 CrossRef CAS PubMed.
- M. Manandhar, K. S. Boulware and R. D. Wood, Gene, 2015, 569, 153–161 CrossRef CAS.
- A. Ciccia, N. Q. McDonald and S. C. West, Annu. Rev. Biochem., 2008, 77, 259–287 CrossRef CAS PubMed.
- W. L. de Laat, E. Appeldoorn, N. G. J. Jaspers and J. H. J. Hoeijmakers, J. Biol. Chem., 1998, 273, 7835–7842 CrossRef CAS PubMed.
- M. Bowles, J. Lally, A. J. Fadden, S. Mouilleron, T. Hammonds and N. Q. McDonald, Nucleic Acids Res., 2012, 40, e101 CrossRef CAS PubMed.
- E. M. McNeil and D. W. Melton, Nucleic Acids Res., 2012, 40, 9990–10004 CrossRef CAS PubMed.
- S. Arora, J. Heyza, H. Zhang, V. Kalman-Maltese, K. Tillison, A. M. Floyd, E. M. Chalfin, G. Bepler and S. M. Patrick, Oncotarget, 2016, 7, 75104–75117 CrossRef PubMed.
- E. M. McNeil, K. R. Astell, A. M. Ritchie, S. Shave, D. R. Houston, P. Bakrania, H. M. Jones, P. Khurana, C. Wallace, T. Chapman, M. A. Wear, M. A. Walkinshaw, B. Saxty and D. W. Melton, DNA Repair, 2015, 31, 19–28 CrossRef CAS PubMed.
- L. P. Jordheim, K. H. Barakat, L. Heinrich-Balard, E. Matera, E. Cros-Perrial, K. Bouledark, R. El Sabeh, R. Perez-Pineiro, D. S. Wishart, R. Cohen, J. Tuszynski and C. Dumontet, Mol. Pharmacol., 2013, 84, 12–24 CrossRef CAS PubMed.
- U. B. Abdullah, J. F. McGouran, S. Brolih, D. Ptchelkine, A. H. El-Sagheer, T. Brown and P. J. McHugh, EMBO J., 2017, 36, 2047–2060 CrossRef CAS.
- T. M. Chapman, C. Wallace, K. J. Gillen, P. Bakrania, P. Khurana, P. J. Coombs, S. Fox, E. A. Bureau, J. Brownlees, D. W. Melton and B. Saxty, Bioorg. Med. Chem. Lett., 2015, 25, 4104–4108 CrossRef CAS PubMed.
- M. R. Hodskinson, J. Silhan, G. P. Crossan, J. I. Garaycoechea, S. Mukherjee, C. M. Johnson, O. D. Schärer and K. J. Patel, Mol. Cell, 2014, 54, 472–484 CrossRef CAS PubMed.
- B. Orelli, T. B. McClendon, O. V. Tsodikov, T. Ellenberger, L. J. Niedernhofer and O. D. Schärer, J. Biol. Chem., 2010, 3705–3712 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9cc05476f |
| ‡ The authors wish it to be known that, in their opinion, the first 3 authors should be regarded as joint first authors. |
| § Present address: Department of Chemistry, Trinity Biomedical Sciences Institute, Trinity College Dublin, Dublin 2, Ireland. |
| This journal is © The Royal Society of Chemistry 2019 |