
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelf-assembly of M4L4 tetrahedral cages incorporating pendant P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/h2_char_e001.gif) S and PSe functionalised ligands†
S and PSe functionalised ligands†
Patrick W. V.
Butler
 a,
Paul E.
Kruger
bc and
Jas S.
Ward
*d
a,
Paul E.
Kruger
bc and
Jas S.
Ward
*d
aResearch School of Chemistry, Australian National University, Acton, Canberra, ACT, Australia
bSchool of Physical and Chemical Sciences, University of Canterbury, Private Bag 4800, Christchurch, 8140, New Zealand
cMacDiarmid Institute for Advanced Materials and Nanotechnology, School of Physical and Chemical Sciences, University of Canterbury, Private Bag 4800, Christchurch 8140, New Zealand
dDepartment of Chemistry, Nanoscience Center, University of Jyväskylä, Jyväskylä 40014, Finland. E-mail: james.s.ward@jyu.fi
First published on 6th August 2019
Abstract
Herein, the synthesis of metal–organic tetrahedral cages featuring flexible thio- and selenophosphate-based ligands is described. The cages were prepared by sub-component self-assembly of A![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) P(OC6H4NH2-4)3 (A = S, Se) or SP(SC6H4NH2-4)3, 2-pyridinecarboxaldehyde, and either Fe[BF4]2 or Co[BF4]2. Preliminary host–guest studies into the ability of the pendant PS and PSe groups to interact with suitable substrates will be discussed.
P(OC6H4NH2-4)3 (A = S, Se) or SP(SC6H4NH2-4)3, 2-pyridinecarboxaldehyde, and either Fe[BF4]2 or Co[BF4]2. Preliminary host–guest studies into the ability of the pendant PS and PSe groups to interact with suitable substrates will be discussed.
The application of metal-directed self-assembly to the formation of discrete architectures has precipitated the development of a myriad of macromolecular metal–organic complexes.1–4 A subset of these, metal–organic cages, are characterised by containing an internal cavity capable of accommodating substrates in host–guest chemistry.5–8 By varying the volume and topology of the internal cavity, substrates from simple organic9–11 and inorganic12,13 molecules to steroids14 and fullerenes15,16 have been successfully encapsulated with those species that most complement the size, shape, bonding and electrostatics of the cavity micro-environment yielding the strongest binding.
The effects of varying the size and shape of the internal cavity are largely intuitive; by contrast, the functionalisation of the micro-environment provides opportunities to investigate more novel host–guest behaviour.17,18 In this regard, the varied and specific non-bonding interactions of main-group functionalities, including dipole–dipole interactions, chalcogen bonding, and hydrogen bonding, are particularly noteworthy and suggests the incorporation of these functionalities into supramolecular cages might facilitate stronger and more selective host–guest chemistry.19,20 Nevertheless, to date main-group functionalised supramolecular cages represent niche structures,21,22 and especially so when considering those with functionalities based on elements beyond the second row of the periodic table.23,24 Inspired by work on phosphorus-based porous organic cages (POCs),25 we have developed a series of metal–organic cages incorporating pendant sulfur and selenium functionalised phosphates.
The basis for the sub-component self-assembly of the cages (Scheme 1) are triamine phosphate pro-ligands, prepared from a mutual tri-substituted phosphite intermediate, subsequently reacted with either elemental sulfur or selenium. The basicity of the amine precluded the direct synthesis of the thio- (1b) and seleno-phosphate (2b) pro-ligands, however, both were found readily accessible via a protecting group strategy (Scheme S1, ESI†). A tetrathiophosphate pro-ligand (3b), analogous to 1b, was similarly prepared with the intention to investigate potential electronic and steric differences compared to the monothiophosphate. To that end, characterisation of 3b demonstrated a significantly altered phosphate environment. In particular, the 31P NMR resonance of the tetrathiophosphate was identified at 98.1 ppm, considerably further downfield relative to 1b (57.5 ppm).
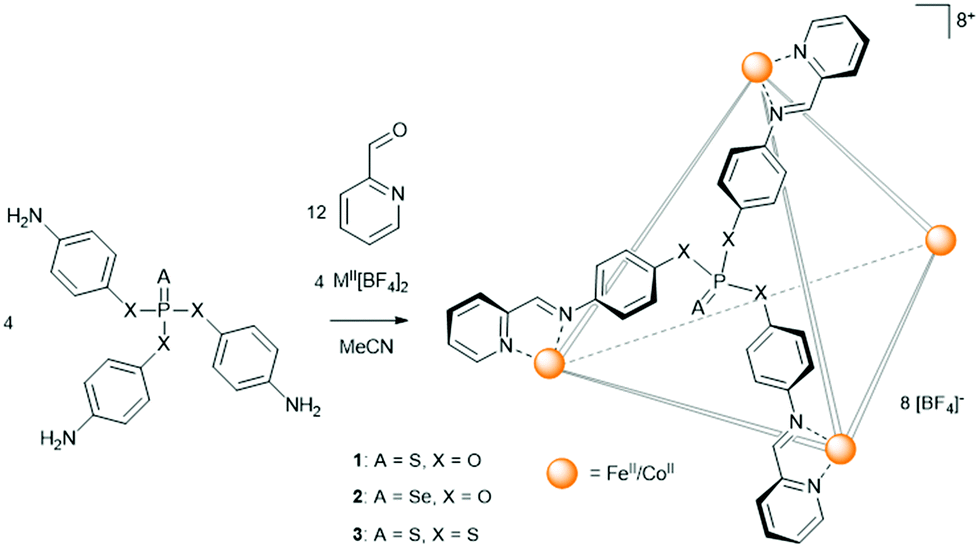 | ||
| Scheme 1 Generalised assembly of the thio- and selenophosphate functionalised M4L4 (M = FeII, CoII) metal–organic cages. | ||
The M4L4 (M = FeII, CoII) face-capped assemblies were prepared by reacting the phosphate pro-ligands (4 equiv.) in acetonitrile with 2-pyridinecarboxaldehyde (12 equiv.) and either Fe[BF4]2 or Co[BF4]2 (4 equiv.).26 Combining the reagents produced immediate colour changes: the Fe reactions became dark purple, a colour characteristic of a low-spin complex, consistent with the strong-field nature of unsubstituted pyridyl-imine ligands,27 while the Co assemblies became yellow. The three Fe4L4 cages, 1c, 2c, and 3c, and the three Co4L4 cages, 1d, 2d, and 3d, were confirmed primarily in solution by HR-ESI MS (Fig. S32–S37, ESI†) with the spectra in each case illustrating a continuous charge series from [M4L4][BF4]53+ to [M4L4]8+. Additional analysis of the isotopic manifolds was found to correlate well with simulated data, supporting the assigned identities of the cages.
Suitable X-ray diffraction quality crystals were obtained for two of the assembled supramolecular cages (1d and 2d), and their solid-state structures elucidated. The molecular structure of 1d (Fig. 1a) revealed the face-capped tetrahedral cage in the orthorhombic P212121 space group, with only the homochiral diastereomer (ΛΛΛΛ) present in the unit cell. Examining the structure it is apparent the thiophosphate ligands have adopted a 3-endo–1-exo configuration, wherein three of the pendant PS groups are positioned inside the internal cavity (endo), while the remaining PS group is positioned outside the cavity (exo). This lopsided arrangement contributes to the relative low symmetry of the structure, which presents a crystallographic asymmetric unit cell containing the entire cage, and in which all the ligands are inequivalent.
 | ||
| Fig. 1 (a) Molecular structures of the thiophosphate-functionalised Co4L4 cage 1d and (b) the selenophosphate-functionalised Co4L4 cage 2d. The volumes of the CoII atoms (orange) are reduced below the van der Waals radii and the hydrogen atoms, solvent molecules, and counterions omitted for clarity. | ||
The separation of the CoII metal nodes of 1d was found to be between 12.6–14.0 Å, which is consistent with the size of similar M4L4 cages reported.23 The cavity volume, however, could not be calculated using various algorithms, and from the space-filling model (Fig. S3, ESI†) it is clear the cavity is largely occupied by the pendant sulfur atoms of the endohedral ligands. The shortest PS⋯SP distance of 3.620(7) Å is very close to the sum of the sulfur–sulfur van der Waals radii (3.60 Å), indicative of a favourable interaction between sulfur atoms within the cage. Despite the small cavity in the solid-state, residual electron density was observed within the cage (8.8 e−), suggesting solvent or water may be able to access the cavity. This would be consistent with the anticipated lability of the ligands in solution to create dynamically larger windows through which guests may ingress or egress.28,29
The three PS groups endo-orientated inside the cage possessed bond lengths within the range 1.917(5)–1.937(7) Å, whilst the final PS group lying in an exo-orientation had a significantly elongated bond length of 2.051(7) Å. The electron density that the exo ligand was interacting with could not be satisfactorily modelled, however, the lengthening of the PS bond is a clear indication that the sulphide groups are not passive in the supramolecular cage.
The molecular structure of 2d (Fig. 1b) was identified in the centrosymmetric C2/c monoclinic space group. Notably, only the achiral diastereomer (ΔΔΛΛ) was present in the unit cell. The cage is of similar size to 1d with Co⋯Co distances ranging from 12.9–14.9 Å; however, in contrast to the thiophosphate cage, each of the pendant PSe groups were found to be orientated endohedrally with respect to the cavity. Given the longer PSe bond length, and the larger atomic size of selenium compared to sulfur, this result infers that the 3-endo–1-exo configuration observed for 1d cannot be attributed to steric overcrowding in the interior of the cage, and further reinforces that the exo PS group is orientated to favourably interact with species outside the cage.
The endo arrangement of the selenophosphate ligands suggests favourable interactions between the pendant PSe groups. These interactions, while able to overcome the associated steric congestion, are non-bonding in nature, with neighbouring Se⋯Se distances of 3.785(3) and 3.811(3) Å beyond the expected length of a formal Se–Se bond (ca. 2.39 Å).30 Furthermore, PSe bond lengths were found to be slightly shorter in the assembly (2.045(6)–2.047(5) Å) compared to the parent pro-ligand (cf. 2.066(1) Å for 2b), strongly indicating the PSe bond is preserved.
The diamagnetic low-spin FeII assemblies, and to a lesser extent the paramagnetic CoII assemblies, permit further characterisation in solution by NMR analysis. Conventional 1D NMR studies of the assemblies (Fig. S25–S30, ESI†) consistently returned complex spectra with 31P NMR analysis indicating 5–7 inequivalent ligand environments for each cage. The clusters of peaks evident in the 1H NMR spectra are characteristic of equilibria between the anticipated cage diastereomers in solution (i.e. homochiral [ΔΔΔΔ/ΛΛΛΛ], heterochiral [ΔΔΔΛ/ΔΛΛΛ] and achiral [ΔΔΛΛ]) in combination with the possible endo/exo configurations (of which five unique configurations exist for M4L4 cages). Such complex NMR spectra are common amongst reported non-templated metal–organic cages and are borne out of the lability in solution of the covalent imine bonds and nitrogen-donor coordination bonds combined with the typically small energy differences between the cage isomers.31,32
Despite the convoluted spectra, diffusion NMR analysis of the Fe assemblies provided strong evidence of the formation of the expected tetrahedral cages (Fig. 2). Analysis of each of the cages found a singular slow diffusing species with estimated diffusion constants from 5.37 × 10−10 to 5.55 × 10−10 m2 s−1, considerably smaller compared to the pro-ligands (13.8 × 10−10 to 14.7 × 10−10 m2 s−1). Furthermore, using the Stokes–Einstein equation,33 calculation of the hydrodynamic diameter of the Fe4L4 cages (23.0 to 23.8 Å) correlated well with the average length between the six vertices for the solid-state structure of 2d (23.6 Å). Considering no other aggregates were observed it can also be concluded that no other thermodynamically stable assemblies resulted from the sub-component self-assembly, and hence the face-capped tetrahedral cages are the sole preferred architecture.
 | ||
| Fig. 2 1H DOSY plot of the thiophosphate functionalised Fe4L4 assembly 1c. The diffusion constant of the assembly was estimated at 5.55 × 10−10 m2 s−1 (black line), whereas the corresponding pro-ligand 1b was estimated at 14.3 × 10−10 m2 s−1 (blue line). | ||
The soft-donor properties of the pendant PS and PSe groups inherently inclines the thio- and selenophosphate functionalised cages to host–guest chemistry with similarly soft guest substrates. A preliminary study with a range of heavy metal ions, including Cu+, Hg2+, Pd2+ and Au+, complementary to both the reduced size and soft bonding of the cavities, consistently found significant changes in the isomer distributions of the cages as measured by 1H and 31P NMR analysis. For example, combining 3c with an excess of [Cu(NCMe3)4][BF4] over 24 h and at 50 °C collapsed the isomer distribution to primarily a single ligand environment (Fig. 3). This isomer collapse is indicative of strong interactions with the dopants, and furthermore consistent with the formation of an energetically favourable host–guest complex.34 Beyond metal ions, the reduced cavity volumes limit the set of potential guests, however, through the modular nature of sub-component self-assembly, options to expand the cages to accommodate larger guests are readily accessible.
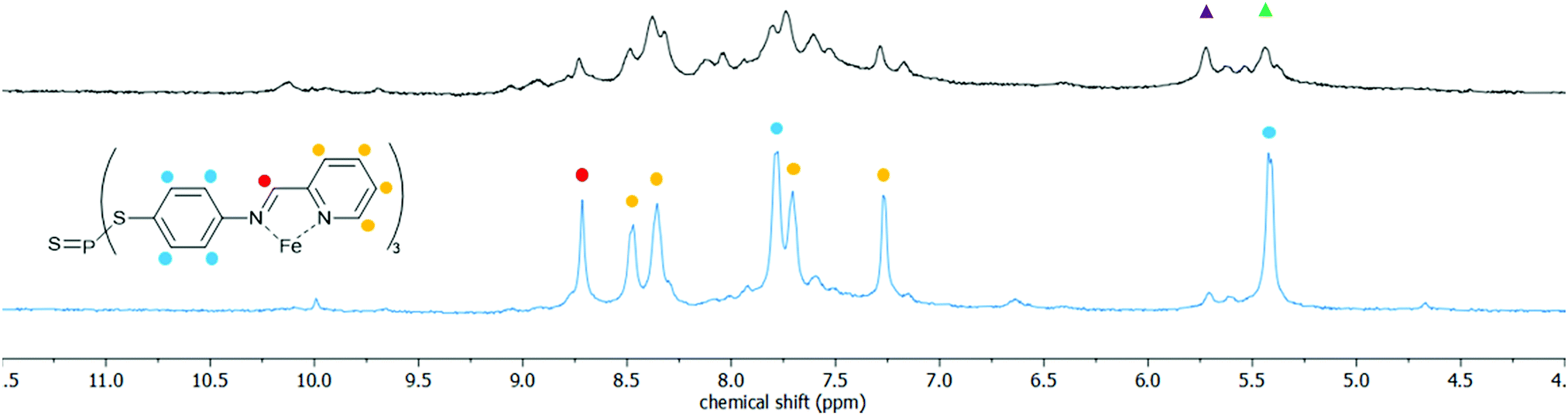 | ||
Fig. 3 The 1H NMR spectrum for the tetrathiophosphate-functionalised Fe4L43c (above) and after mixing with [Cu(NCMe)4][BF4] for 24 h at 50 °C (below). The change in the distribution of the isomers corresponding to the triangle-labelled signals is estimated to be from 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (green:purple) before to 10:1 after. :1 (green:purple) before to 10:1 after. | ||
In conclusion, we have developed a series of related thio- and seleno-phosphate-functionalised metal–organic cages. The face-capped tetrahedral cages were evidenced in solution by HR-MS and diffusion NMR spectroscopy and in the solid-state by single-crystal X-ray diffraction studies. Notably, the phosphate ligands were found to be capable of independently orientating either endohedrally or exohedrally in reference to the internal cavity. Nascent host–guest studies indicated dominant interactions with soft guest substrates, and investigations are ongoing to develop the host–guest chemistry of these soft donor cages.
Conflicts of interest
There are no conflicts to declare.Notes and references
- M. Fujita, Chem. Soc. Rev., 1998, 27, 417 RSC.
- T. R. Cook and P. J. Stang, Chem. Rev., 2015, 115, 7001–7045 CrossRef CAS PubMed.
- M. D. Ward, Chem. Commun., 2009, 4487–4499 RSC.
- D. Fujita, Y. Ueda, S. Sato, H. Yokoyama, N. Mizuno, T. Kumasaka and M. Fujita, Chem, 2016, 1, 91–101 CAS.
- A. Ferguson, R. W. Staniland, C. M. Fitchett, M. A. Squire, B. E. Williamson and P. E. Kruger, Dalton Trans., 2014, 43, 14550–14553 RSC.
- S. J. Dalgarno, N. P. Power and J. L. Atwood, Coord. Chem. Rev., 2008, 252, 825–841 CrossRef CAS.
- H. Amouri, C. Desmarets and J. Moussa, Chem. Rev., 2012, 112, 2015–2041 CrossRef CAS PubMed.
- T. K. Ronson, S. Zarra, S. P. Black and J. R. Nitschke, Chem. Commun., 2013, 49, 2476–2490 RSC.
- W. Cullen, M. C. Misuraca, C. A. Hunter, N. H. Williams and M. D. Ward, Nat. Chem., 2016, 8, 231–236 CrossRef CAS PubMed.
- M. D. Pluth, R. G. Bergman and K. N. Raymond, Acc. Chem. Res., 2009, 42, 1650–1659 CrossRef CAS PubMed.
- G. Szalóki, V. Croué, M. Allain, S. Goeb and M. Sallé, Chem. Commun., 2016, 52, 10012–10015 RSC.
- R. Custelcean, Chem. Soc. Rev., 2014, 43, 1813–1824 RSC.
- H. Amouri, L. Mimassi, M. N. Rager, B. E. Mann, C. Guyard-Duhayon and L. Raehm, Angew. Chem., Int. Ed., 2005, 44, 4543–4546 CrossRef CAS PubMed.
- T. K. Ronson, W. Meng and J. R. Nitschke, J. Am. Chem. Soc., 2017, 139, 9698–9707 CrossRef CAS PubMed.
- K. Mahata, P. D. Frischmann and F. Würthner, J. Am. Chem. Soc., 2013, 135, 15656–15661 CrossRef CAS PubMed.
- M. Yamashina, T. Yuki, Y. Sei, M. Akita and M. Yoshizawa, Chem. – Eur. J., 2015, 21, 4200–4204 CrossRef CAS PubMed.
- P. M. Bogie, L. R. Holloway, C. Ngai, T. F. Miller, D. K. Grewal and R. J. Hooley, Chem. – Eur. J., 2019, 25, 10232–10238 CAS.
- S. Yi, V. Brega, B. Captain and A. E. Kaifer, Chem. Commun., 2012, 48, 10295 RSC.
- M. C. Young, L. R. Holloway, A. M. Johnson and R. J. Hooley, Angew. Chem., Int. Ed., 2014, 53, 9832–9836 CrossRef CAS PubMed.
- R. Custelcean, P. V. Bonnesen, N. C. Duncan, X. Zhang, L. A. Watson, G. Van Berkel, W. B. Parson and B. P. Hay, J. Am. Chem. Soc., 2012, 134, 8525–8534 CrossRef CAS PubMed.
- M. A. Pitt and D. W. Johnson, Chem. Soc. Rev., 2007, 36, 1441 RSC.
- H. W. Roesky, I. Haiduc and N. S. Hosmane, Chem. Rev., 2003, 103, 2579–2596 CrossRef CAS PubMed.
- D. Zhang, T. K. Ronson, J. Mosquera, A. Martinez, L. Guy and J. R. Nitschke, J. Am. Chem. Soc., 2017, 139, 6574–6577 CrossRef CAS PubMed.
- C. T. McTernan, T. K. Ronson and J. R. Nitschke, J. Am. Chem. Soc., 2019, 141, 6837–6842 CrossRef CAS PubMed.
- G. Feng, W. Liu, Y. Peng, B. Zhao, W. Huang and Y. Dai, Chem. Commun., 2016, 52, 9267–9270 RSC.
- A. Ferguson, M. A. Squire, D. Siretanu, D. Mitcov, C. Mathonière, R. Clérac and P. E. Kruger, Chem. Commun., 2013, 49, 1597 RSC.
- E. C. Volpe, P. T. Wolczanski and E. B. Lobkovsky, Organometallics, 2010, 29, 364–377 CrossRef CAS.
- T. K. Ronson, A. B. League, L. Gagliardi, C. J. Cramer and J. R. Nitschke, J. Am. Chem. Soc., 2014, 136, 15615–15624 CrossRef CAS PubMed.
- A. Stephenson, S. P. Argent, T. Riis-Johannessen, I. S. Tidmarsh and M. D. Ward, J. Am. Chem. Soc., 2011, 133, 858–870 CrossRef CAS PubMed.
- B. W. Tattershall, E. L. Sandham and W. Clegg, Dalton Trans., 1997, 81–87 RSC.
- D. Fiedler, D. Pagliero, J. L. Brumaghim, R. G. Bergman and K. N. Raymond, Inorg. Chem., 2004, 43, 846–848 CrossRef CAS PubMed.
- A. M. Castilla, W. J. Ramsay and J. R. Nitschke, Acc. Chem. Res., 2014, 47, 2063–2073 CrossRef CAS PubMed.
- A. Einstein, Ann. Phys., 1905, 322, 549–560 CrossRef.
- J. K. Clegg, J. Cremers, A. J. Hogben, B. Breiner, M. M. J. Smulders, J. D. Thoburn and J. R. Nitschke, Chem. Sci., 2013, 4, 68–76 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental conditions and supporting spectra. CCDC 1938073–1938076. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9cc05443j |
| This journal is © The Royal Society of Chemistry 2019 |