
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
An intermolecular C–H oxidizing strategy to access highly fused carbazole skeletons from simple naphthylamines†
Christian K.
Rank‡
 a,
Alexander W.
Jones‡
b,
Tatjana
Wall
c,
Patrick
Di Martino-Fumo
c,
Sarah
Schröck
c,
Markus
Gerhards
c and
Frederic W.
Patureau
*a
a,
Alexander W.
Jones‡
b,
Tatjana
Wall
c,
Patrick
Di Martino-Fumo
c,
Sarah
Schröck
c,
Markus
Gerhards
c and
Frederic W.
Patureau
*a
aInstitute of Organic Chemistry, RWTH Aachen University, Landoltweg 1, 52074 Aachen, Germany. E-mail: Frederic.patureau@rwth-aachen.de; Web: http://www.patureau-oc-rwth-aachen.de
bFB Chemie, Technische Universität Kaiserslautern, Erwin-Schrödinger Strasse 52, 67663 Kaiserslautern, Germany
cFB Chemie, Technische Universität Kaiserslauter & Research Center Optimas, Erwin-Schrödinger Straße 52, 67663 Kaiserslautern, Germany
First published on 29th October 2019
Abstract
Highly π-extended hetero-cyclic/aromatic skeletons are of great importance as they can be utilized in many organic material based technologies. Therefore, developing efficient, pre-activation-free, synthetic procedures for the rapid build-up of these complex structures remains a high priority objective. The herein presented approach delivers highly fused carbazole skeletons from simple naphthylamine derivatives.
The quest for higher performance aromatic hetero-cyclic fused organic materials imposes increasingly long and tough synthetic routes.1 Indeed, the desired improved properties often demand ever more complicated scaffolds and/or π-extended/fused aromaticity (compounds (I) to (III), Fig. 1).2 In this field, the carbazole backbone is of particular importance, as mentioned in some recent patents on electroluminescent materials.2 The efficient synthesis of these and related structures is therefore a strategic priority. For example, in a recent and inspiring work from Ito & Itami, a highly annulated carbazole structure (2a, Scheme 1) could be accessed from a simple pyrrole (IV) and a (not so trivial) 2,2′-diiodobiphenyl structure (V).3 This previous synthetic method operates through a tetra-fold palladium catalyzed Mizoroki–Heck-like reaction.3 The cost associated to the stoichiometric excess of the precious 2,2′-diiodobiphenyl substrate (IV) however, or any other desired diiodo backbone, significantly limits the applicability and price of the final carbazole based target material. In contrast, oxidative C–H bond activation and functionalization strategies4 remain arguably under-appreciated for accessing such highly fused hetero-cyclic π-extended novel materials, in spite of seminal oxidation studies in the middle of the 20th century.5 Moreover, inspiring work by Hiroto and Shinokubo showed recently that primary anthramines such as (VI) could easily undergo oxidation with DDQ (2,3-dichloro-5,6-dicyano-1,4-benzoquinone) to deliver the corresponding annulated carbazole skeleton ((VII), Scheme 1).6 Unfortunately, the latter method is limited to very electron rich primary anthramine starting materials. These alternative C–H functionalization strategies are nevertheless essential in order to make the future technologies based on these materials as affordable as possible. We therefore got interested in Ito and Itami's highly annulated carbazole 2a (Scheme 1), and attempted a more direct C–H oxidative functionalization approach. For this purpose, we inspired ourselves from the historical reports describing the more challenging direct oxidation of N-phenyl-2-naphthylamine, wherein the yield of corresponding annulated carbazole 2b does not exceed 25%.5 We thus foresaw that the latter process might be improved by optimizing the oxidizing method, efficiently affording these valuables hetero-cyclic/aromatic skeletons, without pre-activation, from trivial building blocks.
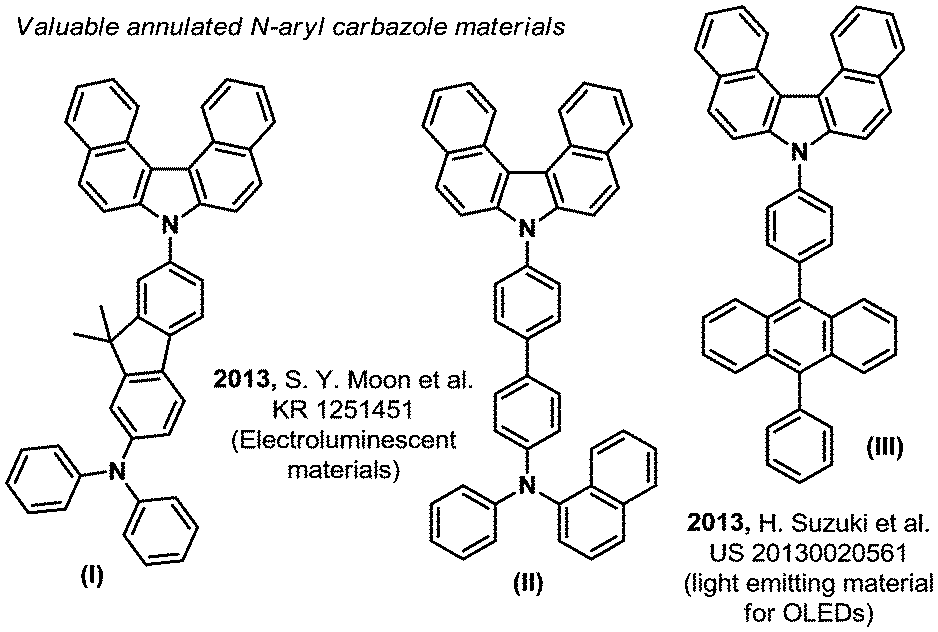 | ||
| Fig. 1 Some N-aryl-dibenzo-carbazole derived electroluminescent materials. | ||
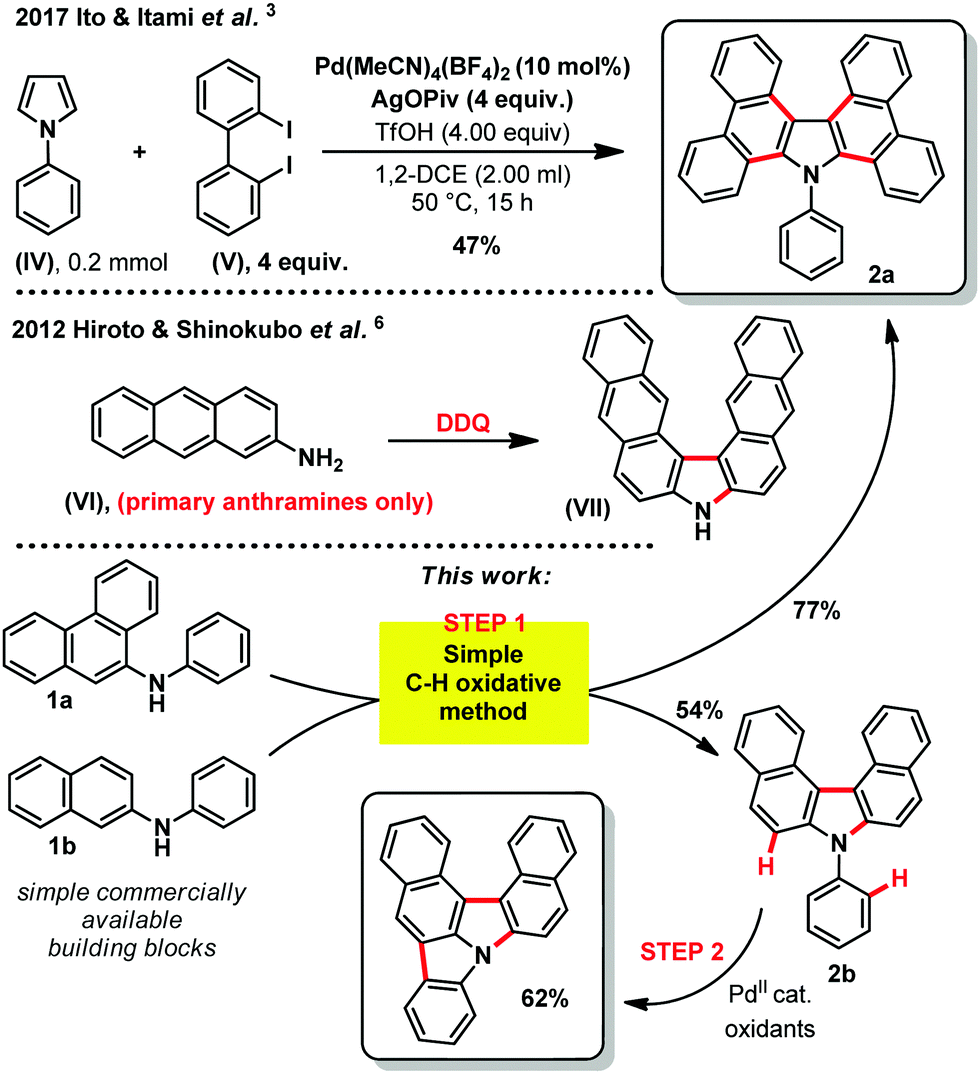 | ||
| Scheme 1 Strategies for accessing fused carbazole skeletons from naphthylamine derivatives. | ||
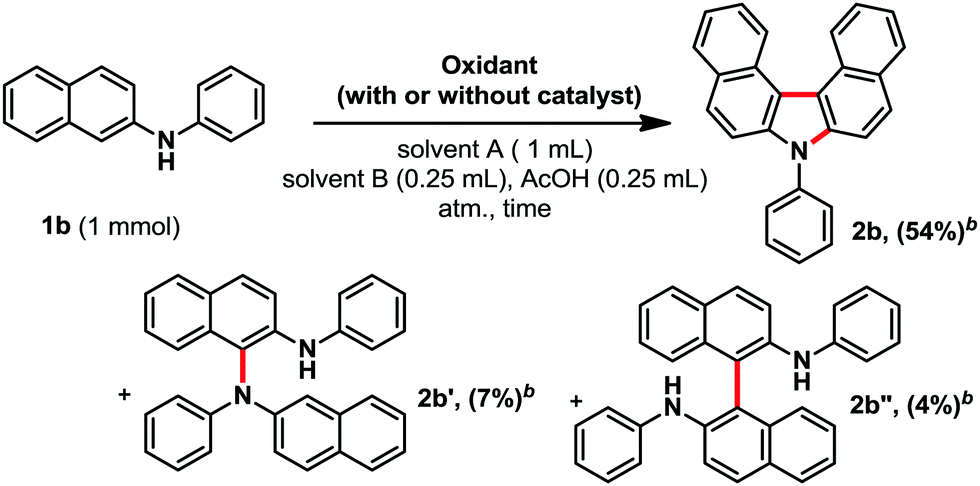
While the historical reports focused on potassium permanganate, or peroxides as oxidants, yielding usually only traces of product 2b,5 we turned our attention to silver oxide. The latter is a mild, easy to handle, and relatively basic oxidant, and is therefore well suited for electron-rich and sensitive heterocycles. In addition, silver oxide is one of the cheapest AgI salt available. Moreover, in most cases, the reduced silver particles usually precipitate, leading to an easy recovery of the metal through simple filtration. It should otherwise be noted that the reaction does not need any other precious metal catalyst. Elements of optimization are shown in Table 1. Next, we also optimized the solvent system and reaction time (see ESI†). We eventually settled for a mixture of toluene, cumene and acetic acid (halide-free system), for a reaction time of two hours, affording product 2b in 54% isolated yield (Table 1, entry 9). Interestingly, in those conditions two minor side products 2b′ (7%) and 2b′′ (4%) could be isolated and identified (Table 1). From these, we assume that this reaction could follow a radical pathway.5b Moreover, TEMPO, a classical radical scavenger, poisons the reaction by either trapping reactive radical intermediates and/or neutralizing the silver oxidant (2b, 0%, entry 5). It should be noted that changing the acid to PivOH, CF3CO2H, or a mixture of AcOH and NaOAc, did not improve the reaction. Moreover, the addition of any other base led to a significant decrease in conversion towards product 2b (see ESI†). Importantly, any other silver salt or the addition of cationic silver sources to Ag2O did not have a positive influence on the reaction outcome (see ESI†).
|
|
||||
|---|---|---|---|---|
| # | Catalyst or oxidant | [mmol] | Atm. [1 bar] | Yielda (%) |
| a All reactions were carried out using 1b (1 mmol), oxidant, in C2Cl4 (1 mL), PhCl (0.25 mL), AcOH (0.25 mL), at 60 °C for 24 h. The yield was determined by 1H-NMR analysis of the crude reaction mixture using 1,2-DCE (1,2-dichloroethane) as an internal standard. b Isolated yields in brackets. c 150 °C. d C2Cl4 and PhCl are replaced by toluene (1 mL) and cumene (0.25 mL), reaction time is reduced to 2 h (see ESI for details). | ||||
| 1c | [Ru(C6Me6)Cl2]2 + Cu(OAc)2 | 0.05 1.10 | O2 | 14 |
| 2 | Ag 2 O | 0.50 | Air | 42 |
| 3 | Ag 2 O | 0.75 | Air | 44 |
| 4 | Ag 2 O | 1.00 | Air | 50 |
| 5 | Ag 2 O + 1 equiv. TEMPO | 1.00 | Air | 0 |
| 6 | Ag 2 O | 1.00 | O2 | 47 |
| 7 | Ag 2 O | 1.00 | N2 | 50 |
| 8 | Ag 2 O | 1.25 | Air | 50 |
| 9d | Ag 2 O | 1.00 | Air | 56(54) |
With the optimized conditions in hand, we then explored the scope of the reaction (Scheme 2). It was possible to form several highly π-extended carbazole derivatives in good yields (for such motifs), and with some functional group tolerance. Importantly, Ito and Itami's highly annulated carbazole product 2a was isolated with a 77% yield, significantly increasing the previous state-of-the-art yield for this motif (Schemes 1 and 2). Next, we were curious whether we might be able to perform the highly strained dehydrogenative ring closure of the π-extended N-aryl-carbazole structure, towards the highly fused indolcarbazole skeleton 3 (nomenclature name: indolo[3.2.1-jk]carbazole), with potentially interesting properties.7,8 To the best of our knowledge however, dehydrogenative Scholl/Mallory type ring closing systems9 have not (yet) been found competent for this type of scaffold. We therefore adapted a palladium catalysed oxidative C–H bond activation method by Patureau.8 Satisfyingly, we were able to obtain compound 3b in 62% isolated yield (Scheme 3), which represents a fairly good yield for this sort of dehydrogenative synthetic method.8 No dehydrogenative cyclization product with substrate 2a was observed however.
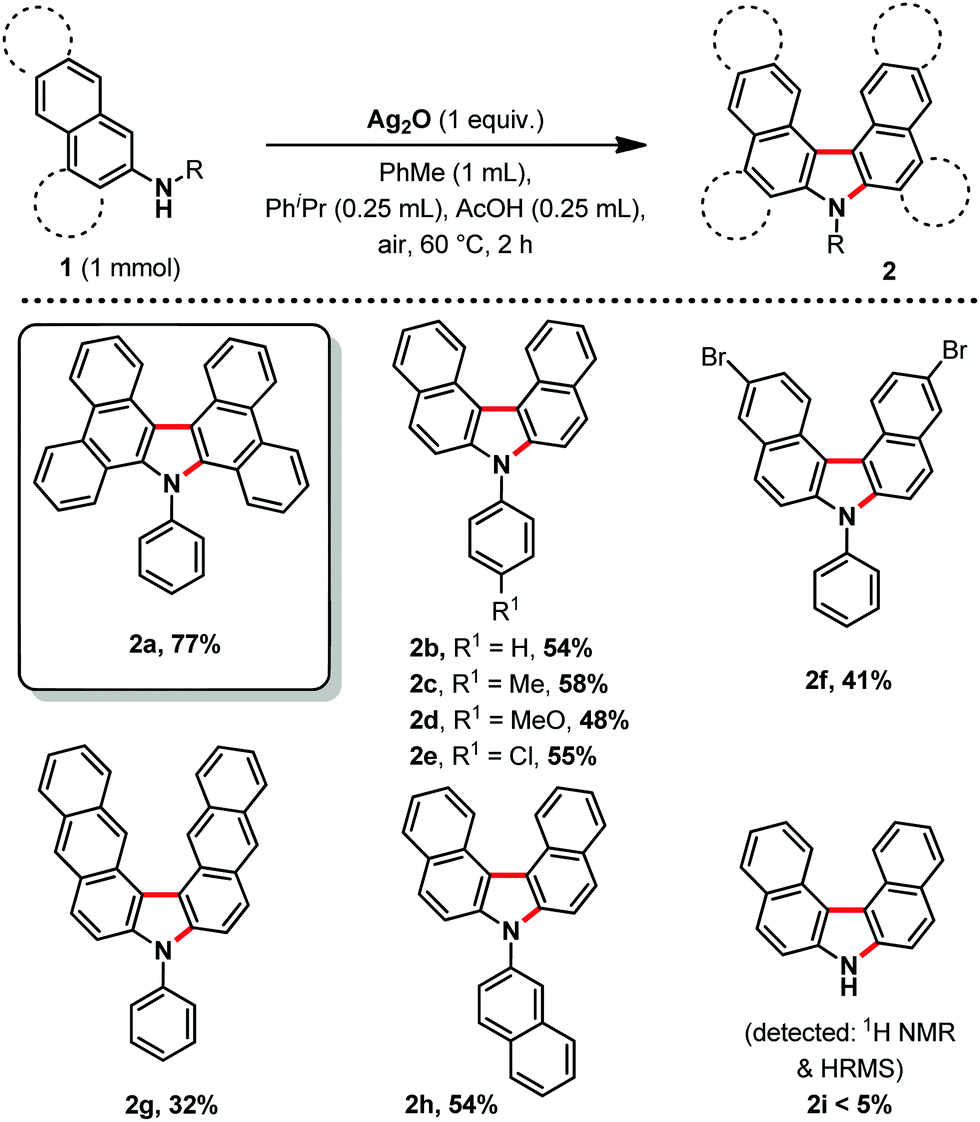 | ||
| Scheme 2 Reaction scope, isolated yields, all reactions were carried out using 1 (1.00 mmol), Ag2O (1.00 equiv.), PhMe (1.00 mL), PhiPr (0.25 mL), AcOH (0.25 mL), under air at 60 °C for 2 h. | ||
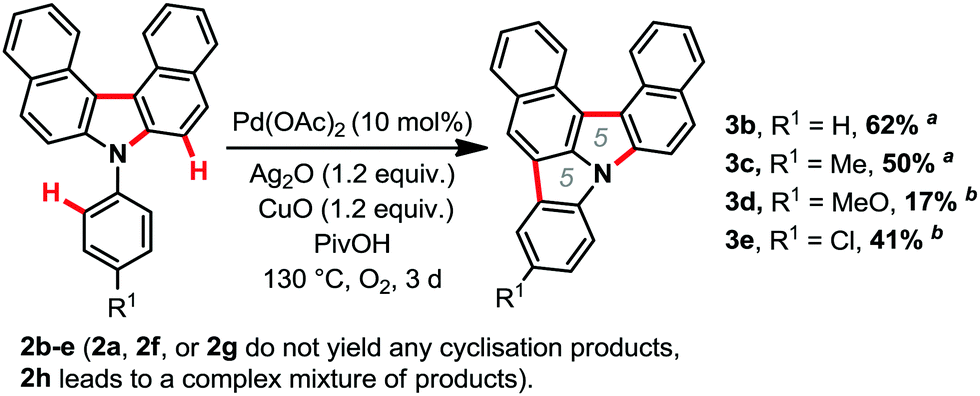 | ||
| Scheme 3 Palladium catalysed strained dehydrogenative ring closure,8 isolated yields. a0.5 mmol scale, PivOH: 1.5 mL. b0.12 mmol scale. | ||
Thereafter, we looked at all these novel molecules using DFT calculations10–15 (see ESI†) as well as UV and fluorescence spectroscopy. Importantly, the DFT calculations show a significant increase of the planarity from backbone 2b to 3b, by about a third (torsion angle from −27° to −18°, respectively, see DFT data in the ESI†). This is due to an enlargement of the gulf between the two naphthyl moieties in 3b, allowing for a more planar disposition. The electronic properties of the compounds moreover differ from scaffold 2 to the fused π-system of scaffold 3. The observed transitions show a π → π* character. As displayed in the figure for highly fused product 3b, the LUMO delocalizes over the entire molecule. In contrast, the HOMO seems to be localized only on the upper carbazole unit (Fig. 2 and 3). The highly fused product 3b displays a broad absorption band between 250 and 400 nm. Moreover, an emission spectrum (λEX = 270 nm) with a maximum emission wave at 428 nm was obtained.
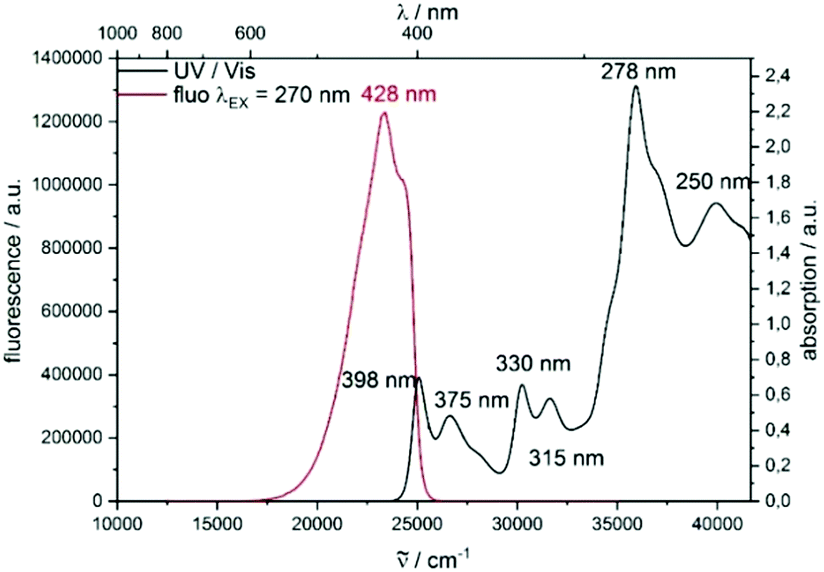 | ||
| Fig. 2 Normalized UV/vis absorption and emission spectra of 3b in dichloromethane. | ||
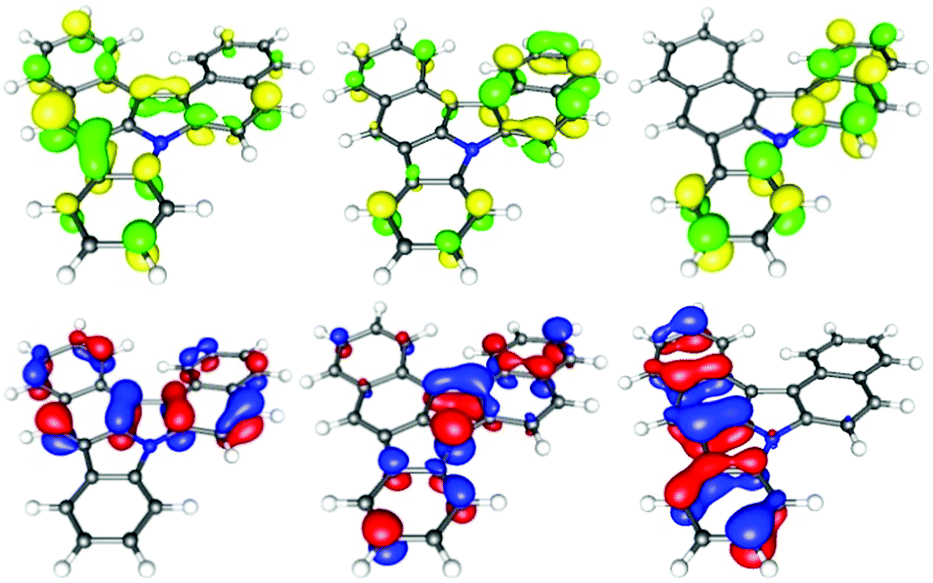 | ||
| Fig. 3 Frontier molecular orbitals of 3b (LUMO (3.38 eV), LUMO+1 (4.12 eV) and +2 (4.75 eV), from left to right, top, and HOMO (0 eV), HOMO+1 (−0.26 eV) and +2 (−0.83 eV), from left to right, beneath). | ||
Finally, the quantum yield was determined for the two model systems 2b and 3b (see Table 2). The determined values are generally between 0.28 and 0.45, depending on the excitation wavelength. A clear deviation of the additionally annulated system could not be observed. The second cyclization (3b) only shows a little influence on the quantum yield. Moreover, these values are somewhat lower compared to other recently appeared fused heterocycles.2d
| Entry | Substance/wave number | Standard | Quantum yield | Confidence |
|---|---|---|---|---|
| 1 | 2b/366 nm | POPOP | 0.33 | ±0.03 |
| 2 | 2b/350 nm | POPOP | 0.45 | ±0.03 |
| 3 | 2b/280 nm | Tryptophan | 0.43 | ±0.03 |
| 4 | 3b/375 nm | POPOP | 0.43 | ±0.03 |
| 5 | 3b/330 nm | POPOP | 0.36 | ±0.02 |
| 6 | 3b/277 nm | Tryptophan | 0.28 | ±0.03 |
In summary, we developed an efficient route to synthesize dibenzocarbazole as well as highly fused π-extended dibenzo-indolocarbazole derivatives, utilizing only simple oxidative C–H functionalization conditions. In general, the utilization of simple oxidative C–H bond functionalization strategies can be expected to rise in the next few years. The fluorescence quantum yields are a further aspect for optimization.
DFG-funded transregional collaborative research center SFB/TRR 88 “Cooperative effects in homo and heterometallic complexes” (http://3MET.de, B10 in cooperation with C2), DFG funded project PA 2395/2-1, COST Action CA15106 (CHAOS), and ERC project 716136: “2O2ACTIVATION,” are acknowledged for financial support.
Conflicts of interest
There are no conflicts to declare.Notes and references
- For selected reviews, see: (a) W. Wu, Y. Liu and D. Zhu, Chem. Soc. Rev., 2010, 39, 1489 RSC; (b) A. Narita, X.-Y. Wang, X. Feng and K. Muellen, Chem. Soc. Rev., 2015, 44, 6616 RSC; (c) M. Stepien, E. Gonka, M. Zyla and N. Sprutta, Chem. Rev., 2017, 117, 3479 CrossRef CAS PubMed; (d) M. Krzeszewski, D. Gryko and D. T. Gryko, Acc. Chem. Res., 2017, 50, 2334 CrossRef CAS PubMed; (e) H. Zhylitskaya and M. Stępień, Org. Chem. Front., 2018, 5, 2395 RSC; (f) T. Janosik, A. Rannug, U. Rannug, N. Wahlström, J. Slätt and J. Bergman, Chem. Rev., 2018, 118, 9058 CrossRef CAS PubMed.
- (a) T. Ishisone, S. Seo, Y. Nonaka, T. Kawata and N. Ohsawa, J. Soc. Inf. Disp., 2014, 22, 404 CrossRef CAS; (b) S. Y. Moon, B. S. Lee, S. H. Lee, G. M. Lee, D. H. Choi, D. H. Kim and J. H. Park, Preparation of carbazoles for organic electronic element, KR Pat., 2013106255, 2013 Search PubMed; (c) H. Suzuki, S. Seo, S. Kawakami and T. Suzuki, Dibenzo[c,g]carbazole compounds, light-emitting components, light-emitting devices, display devices, lighting devices and electronic devices, US Pat., 20130020561, 2013 Search PubMed; (d) X. Pu, M. Zhang, J. Lan, S. Chen, Z. Liu, W. Liang, Y. Yang, M. Zhang and J. You, Org. Lett., 2019, 14, 1139 CrossRef PubMed.
- (a) W. Matsuoka, H. Ito and K. Itami, Angew. Chem., Int. Ed., 2017, 56, 12224 CrossRef CAS PubMed; (b) H. Kitano, W. Matsuoka, H. Ito and K. Itami, Chem. Sci., 2018, 9, 7556 RSC.
- For some selected reviews, see: (a) C.-L. Sun, B.-J. Li and Z.-J. Shi, Chem. Commun., 2010, 46, 677 RSC; (b) C. S. Yeung and V. M. Dong, Chem. Rev., 2011, 111, 1215 CrossRef CAS PubMed; (c) L. McMurray, F. O’Hara and M. J. Gaunt, Chem. Soc. Rev., 2011, 40, 1885 RSC; (d) P. B. Arockiam, C. Bruneau and P. H. Dixneuf, Chem. Rev., 2012, 112, 5879 CrossRef CAS PubMed; (e) J. Yamaguchi, A. D. Yamaguchi and K. Itami, Angew. Chem., Int. Ed., 2012, 51, 8960 CrossRef CAS PubMed; (f) S. I. Kozhushkov and L. Ackermann, Chem. Sci., 2013, 4, 886 RSC; (g) C. Liu, J. Yuan, M. Gao, S. Tang, W. Li, R. Shi and A. Lei, Chem. Rev., 2015, 115, 12138 CrossRef CAS PubMed; (h) T. Gensch, M. N. Hopkinson, F. Glorius and J. Wencel-Delord, Chem. Soc. Rev., 2016, 45, 2900 RSC; (i) Y. Qin, L. Zhu and S. Luo, Chem. Rev., 2017, 117, 9433 CrossRef CAS PubMed; (j) D.-S. Kim, W.-J. Park and C.-H. Jun, Chem. Rev., 2017, 117, 8977 CrossRef CAS PubMed.
- Early studies on the direct oxidation of N-phenyl-2-napthylamine: (a) J. Rehner Jr., F. W. Banes and S. B. Robison, J. Am. Chem. Soc., 1945, 67, 605 CrossRef; (b) D. F. Bowman, B. S. Middleton and K. U. Ingold, J. Org. Chem., 1969, 34, 3456 CrossRef CAS; (c) M. Smrcina, S. Vyskocil, B. Maca, M. Polasek, T. A. Claxton, A. P. Abbott and P. Kocovsky, J. Org. Chem., 1994, 59, 2156 CrossRef CAS . For related works, see: ; (d) K. Nakano, Y. Hidehira, K. Takahashi, T. Hiyama and K. Nozaki, Angew. Chem., Int. Ed., 2005, 44, 7136 CrossRef CAS PubMed; (e) N. Ogawa, Y. Yamaoka, H. Takikawa, K. Tsubaki and K. Takasu, J. Org. Chem., 2018, 83, 7994 CrossRef CAS PubMed.
- K. Goto, R. Yamaguchi, S. Hiroto, H. Ueno, T. Kawai and H. Shinokubo, Angew. Chem., Int. Ed., 2012, 51, 10333 CrossRef CAS PubMed.
- Pre-activated route: (a) H. G. Dunlop and S. H. Tucker, J. Chem. Soc., 1939, 1945 RSC; (b) C. Niebel, V. Lokshin, A. Ben-Asuly, W. Marine, A. Karapetyanac and V. Khodorkovsky, New J. Chem., 2010, 34, 1243 RSC; (c) M. Rivoal, L. Bekere, D. Gachet, V. Lokshin, W. Marine and V. Khodorkovsky, Tetrahedron, 2013, 69, 3302 CrossRef CAS; (d) S. I. Wharton, J. B. Henry, H. McNab and A. R. Mount, Chem. – Eur. J., 2009, 15, 5482 CrossRef CAS PubMed; (e) J. Lv, Q. Liu, J. Tang, F. Perdih and K. Kranjc, Tetrahedron Lett., 2012, 53, 5248 CrossRef CAS; (f) P. Kautny, D. Lumpi, Y. Wang, A. Tissot, J. Bintinger, E. Horkel, B. Stoeger, C. Hametner, H. Hagemann, D. Mab and J. Froehlich, J. Mater. Chem. C, 2014, 2, 2069 RSC.
- Dehydrogenative route: A. W. Jones, M.-L. Louillat-Habermeyer and F. W. Patureau, Adv. Synth. Catal., 2015, 357, 945 CrossRef CAS.
- Scholl-type/Mallory-type reactions, see for example: (a) M. Grzybowski, K. Skonieczny, H. Butenschoen and D. T. Gryko, Angew. Chem., Int. Ed., 2013, 52, 9900 CrossRef CAS PubMed; (b) F. B. Mallory and C. W. Mallory, Org. React., 1984, 30, 1 CAS; (c) W. H. Laarhoven, Org. Photochem., 1989, 10, 163 CAS; (d) H. Meier, Angew. Chem., Int. Ed. Engl., 1992, 31, 1399 CrossRef; (e) Y. Tominaga and R. N. J. Castle, Heterocycl. Chem., 1996, 33, 523 CrossRef CAS; (f) S. Hagen and H. Hopf, Top. Curr. Chem., 1998, 196, 45 CrossRef CAS; (g) K. B. Jørgensen, Molecules, 2010, 15, 4334 CrossRef PubMed.
- Methods of Quantum Chemical Calculations: Input structures for the compounds were generated by applying the universal force field (UFF)11 implemented in the Avogadro program.12 We performed structure optimization in Gaussian 0913 with the (TD)-DFT functional B3LYP as implemented in Turbomole 7.314 by addition of Grimme's dispersion correction D3.15 The def2-TZVP basis set was used for all elements. Optimized structures were tested for minimum geometry by application of frequency calculations. No imaginary frequencies were found. The scaling factors 0.956 (CH stretching modes) and 0.97 (fingerprint) were used. To calculate the vertical electronic transitions, TD-DFT was used with the previously mentioned functional and basis set.
- A. K. Rappe, C. J. Casewit, K. S. Colwell, W. A. Goddard and W. M. Skiff, J. Am. Chem. Soc., 1992, 114, 10024 CrossRef CAS.
- M. D. Hanwell, D. E. Curtis, D. C. Lonie, T. Vandermeersch, E. Zurek and G. R. Hutchison, J. Cheminf., 2012, 4, 17 CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 09, Revision A.02, Gaussian, Inc., Wallingford, CT, 2016 Search PubMed.
- F. Furche, R. Ahlrichs, C. Hättig, W. Klopper, M. Sierka and F. Weigend, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2014, 4, 91 CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures and analytical data. See DOI: 10.1039/c9cc05240b |
| ‡ C. K. Rank and A. W. Jones contributed equally to this work and are considered co-first authors. This work was started at the Technische Universität Kaiserslautern and finished at the RWTH Aachen University. |
| This journal is © The Royal Society of Chemistry 2019 |