
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Gel to gel transitions by dynamic self-assembly†
Santanu
Panja
 and
Dave J.
Adams
*
and
Dave J.
Adams
*
School of Chemistry, University of Glasgow, Glasgow, G12 8QQ, UK. E-mail: dave.adams@glasgow.ac.uk
First published on 2nd August 2019
Abstract
Dynamic systems are of great interest from the perspective of mimicking biology through to preparing useful and exciting materials. Transient supramolecular gels are potentially useful, but there are limited applications that require a gel that only exists for a short length of time. Here, we show how a dynamic system can be designed to prepare materials with properties that cannot be directly accessed using the same gelator.
Gels are important and useful materials, both academically and industrially. These materials contain significant amounts of liquid, but behave as solids. The solid-like behaviour arises from a network, which immobilises the solvent. The networks can be essentially permanent (as in cross-linked polymer gels). In comparison, supramolecular gels are formed by networks that are a result of non-covalent interactions; as a result, these networks can be readily disassembled, resulting in a return to the solution state.1–4
Typically, gels are formed and then used, generally with the aim that their mechanical properties do not change over their useful lifetime. However, dynamic systems are becoming increasingly of interest.5–15 In these dynamic gel systems, typically a supramolecular gel is formed on application of an energy input or fuel, which drives the formation of a molecule capable of forming a gel.5,12–14,16–19,20 When the energy source is turned off or the fuel runs out, the system returns to its original, non-gelling state. Hence, transient gels can be formed, the duration of which is controlled by the concentration of the fuel or energy inputted. Such gels are interesting, and there have been a small number of suggested applications.7 As a specific example describing a change in state from a solution to a gel phase, followed by a return to the solution phase when the fuel runs out or energy input ceases, Tena-Solsona et al. have shown that a transient gel can be formed using the addition of a fuel to a Fmoc-amino acid, which can be used to form self-erasing inks.18 However, it has been pointed out that most (if not all) of the dynamic assemblies reported so far have no practical application.21 For the field to progress, this needs to change.
Another interesting, but as yet unrealised, possibility is a reconfiguring of state to give a different material at the end point, for example a change from a solution to a gel, followed by formation of a different type of gel network (Fig. 1). This could be an extremely powerful approach to adapt material properties, annealing the properties, controlling diffusion or synergistic catalytic effects by changing the network for example. It may also be possible to access materials which cannot be formed directly. This could be potentially be achieved in a spatially controlled manner.
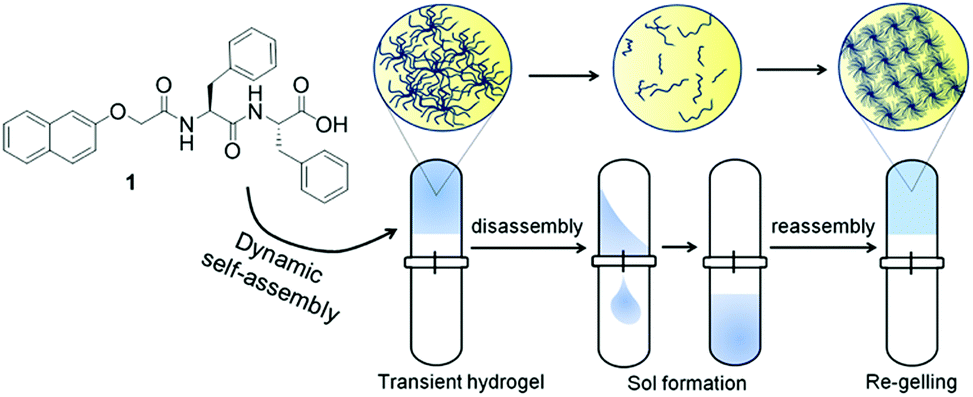 | ||
| Fig. 1 Inducing gel to gel transitions by dissipative assembly. The gelator used here 1 can be used to form a transient gel, with disassembly driven by an increase in pH. At high pH, a second type of gel network is formed. | ||
When a DMSO solution of 1 (Fig. 1) is diluted with H2O (final ratio 20/80 DMSO/water (v/v)), phase separation occurs and a self-supporting gel is formed at a concentration of 1 of 2 mg mL−1 (Fig. S1, ESI†).22 The gel consists of densely packed spherulitic domains of fibres as observed from confocal microscopy imaging (Fig. S2, ESI†). Rheological studies showed that the storage modulus (G′) is significantly higher than the corresponding loss modulus (G′′) and both are independent of frequency (Fig. S3 and S4, ESI†). The gel has strain bearing capacity of ∼5% before it started to collapse. The pH of the gel was measured to be ∼4.1. A stable gel was also formed when urease is present, or when urease and a calcium salt is present (Fig. S1, ESI†), with the gels having very similar microstructure (Fig. S2, ESI†). The rheological moduli are slightly lower in the presence of a calcium salt and/or the enzyme (Fig. S3 and S4, ESI†).
At this ratio of DMSO and H2O, 1 exhibits a pKa of 5.7 (Fig. S5, ESI†); therefore, if we increase the pH of the gel above a pH 5.7, the gel would be expected to return into a solution state.23 This is indeed the case if we adjust the pH with sodium hydroxide (Fig. S6, ESI†). Gel formation does not occur if the initial pH is above 5.7 (Fig. S7, ESI†).
As a first step towards constructing an out-of-equilibrium system involving the DMSO/water gel of 1, we used a slow increase in pH as a counter trigger to drive the self-assembly towards non-assembled states. The autocatalytic reaction between urease and urea produces NH313,24,25 that can trigger homogeneous hydrogelation of cationic amphiphiles at high pH.26 We therefore examined the rate of pH change involving urease–urea reaction in presence of gelator 1. When an aqueous solution of the enzyme was added to the vial containing a mixture of urea (in water) and gelator (in DMSO), initially the pH dropped to ∼pH 4.1 and a gel was formed as above (Fig. 2).
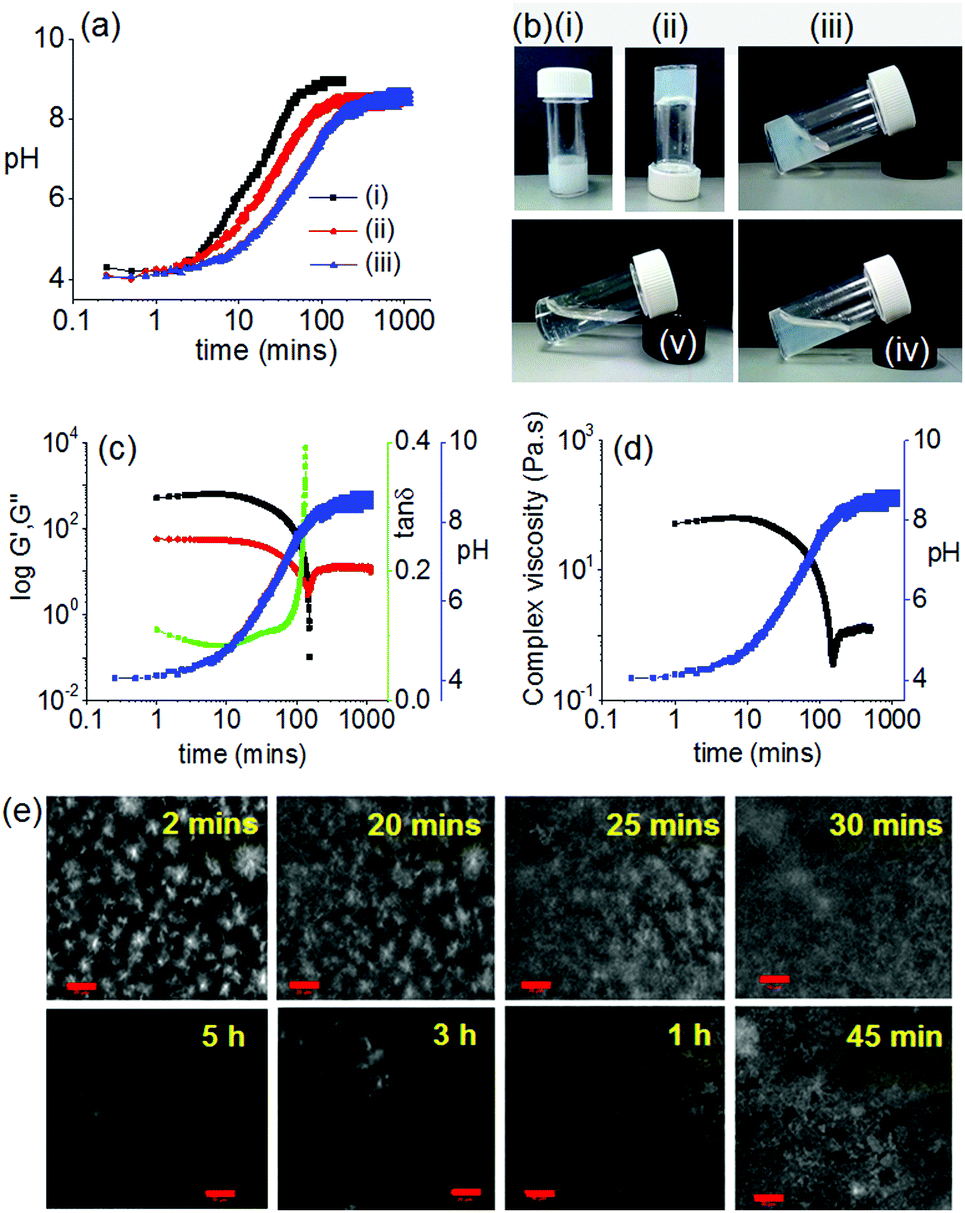 | ||
Fig. 2 Transient gel formation by addition of urease. (a) Change in pH with time from the urease–urea reaction in presence of 1. Initial reaction conditions are (i) [urease] = 0.2 mg mL−1, [urea] = 0.02 M; (ii) [urease] = 0.1 mg mL−1, [urea] = 0.02 M; (iii) [urease] = 0.1 mg mL−1, [urea] = 0.01 M. (b) Photographs showing the phase change with time when aqueous solution of the enzyme was added to the mixture of 1 in DMSO and urea: (i) immediately after addition, (ii) after 2 minutes, (iii) after 30 minutes, (iv) after 1 hour, (v) after 16 hours. (c) Variation of G′ (black), G′′ (red), tan![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) δ (green) and pH (blue) with time for 1 in presence of urea–urease reaction. (d) Variation of complex viscosity (black) and pH (blue), with time for 1 in presence of urea–urease reaction. (e) Time dependent confocal microscopy images of 1 in presence of urea–urease reaction (scale bars represent 20 μm). For (a)–(e), solvent is 20/80 DMSO/water (v/v) and [1] = 2 mg mL−1. For (b)–(e), Initial [urease] = 0.1 mg mL−1 and [urea] = 0.01 M. δ (green) and pH (blue) with time for 1 in presence of urea–urease reaction. (d) Variation of complex viscosity (black) and pH (blue), with time for 1 in presence of urea–urease reaction. (e) Time dependent confocal microscopy images of 1 in presence of urea–urease reaction (scale bars represent 20 μm). For (a)–(e), solvent is 20/80 DMSO/water (v/v) and [1] = 2 mg mL−1. For (b)–(e), Initial [urease] = 0.1 mg mL−1 and [urea] = 0.01 M. | ||
With time, the pH of the medium increased slowly and reached above pH 5.7 (pKa of 1) within ∼30 min (Fig. 2a). As expected, at a pH above the pKa, the gel was not stable, but rather a highly viscous material was formed over time which did not allow the inversion of vial. The viscous material progressively converted to a clear solution with time (Fig. 2b). Interestingly, the final viscosity of the solution is higher than if a solution is prepared directly at high pH (Fig. S8, ESI†). Hence, as expected, this method allows the preparation of gels with a transient lifetime. The lifetime of the gel could be easily controlled by the concentration of the enzyme and urea (Fig. S9 and S10, ESI†).
The resulting non-equilibrium assemblies can be further understood using time sweep rheology (Fig. 2c), viscosity (Fig. 2d) and confocal microscopy (Fig. 2e). From the rheology, initially the storage modulus (G′) was considerably higher than the loss modulus (G′′), showing that a gel is formed quickly on addition of water to the DMSO solution of 1. As the pH of the system increases, G′ begins to decrease indicating destruction of the intermolecular associations. The rate of pH change can be controlled by varying the concentration of urease or urea (Fig. 2a). When the rate of pH change was slow, the decrease in G′ was significantly delayed as compared to a system where the rate of pH change was higher (Fig. S9, ESI†). In both cases, initially tanδ (G′′/G′) decreases and becomes almost constant for a limited time frame before it started to increase again. The viscosity data also depends on pH and mirrors the rheology. Time dependent confocal microscopy imaging was used to show the presence of spherulitic domains of fibres initially (as expected from this solvent-triggered approach22). With time, these domains become less distinct and disappear, leading to the gel converting to a solution (Fig. 2e and Fig. S9, ESI†). We note that there was not a significant change in the viscosity of the final solutions irrespective of the initial concentrations of urea and enzyme (Fig. S11, ESI†).
Homogeneous hydrogelation of solutions of 1 directly at high pH can be achieved in presence of Ca2+ ions.22 The divalent ion cross-links the worm-like micelles that are formed at high pH. With this in mind, we incorporated a calcium salt in our systems, initially at a concentration of 0.5 molar equivalents with respect to 1. As mentioned above, there is little effect on the initially-formed gels. In the presence of urease and urea, the pH again increased with time (Fig. 3a and Fig. S12, ESI†). The rate of pH change was again dependent on the concentration of the urease and urea; the rate was also dependent on the concentration of Ca2+ (Fig. S12, ESI†). At higher concentrations of Ca2+, a slight decrease in pH is observed at longer times. It is not currently clear why this occurs. Under all conditions, instead to the formation of solutions as in the absence of Ca2+, at high pH we obtained gels that were transparent in comparison to the DMSO–H2O gel (Fig. 3b and Fig. S13, ESI†). Hence, the dissipative assembly here leads to an unusual gel-to-sol-to-gel transition.
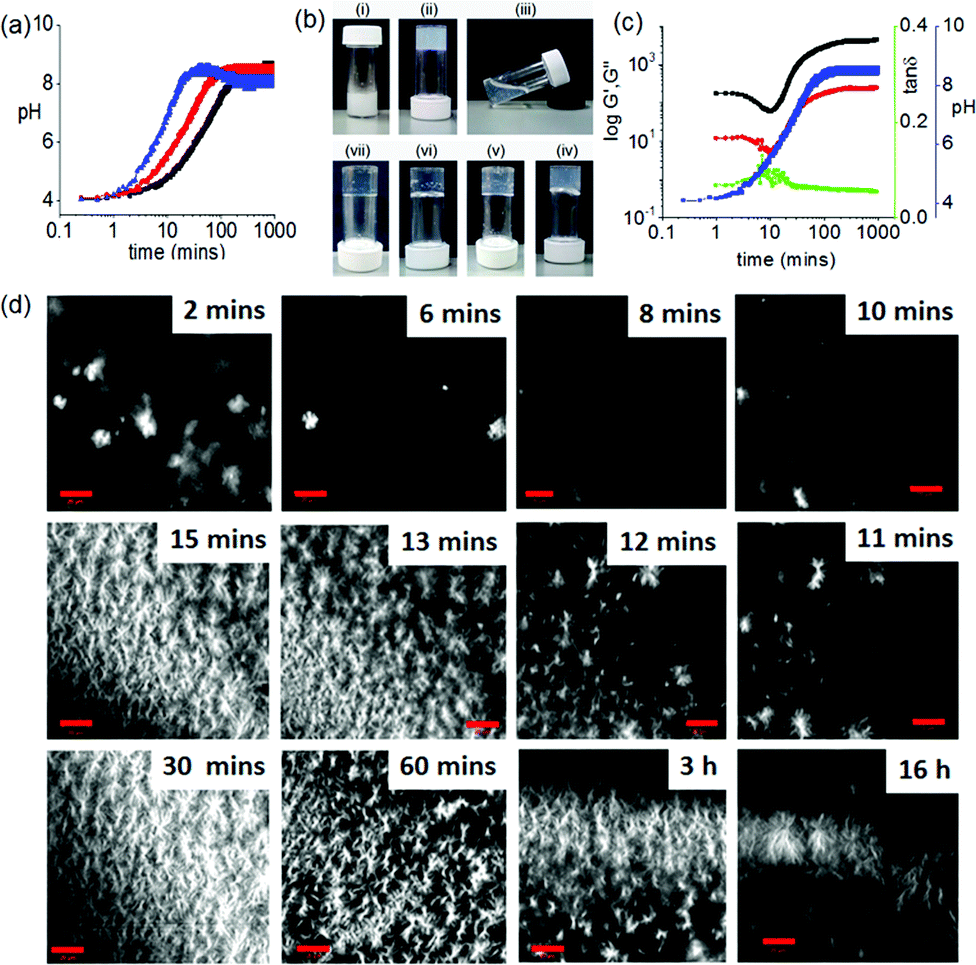 | ||
| Fig. 3 Homogeneous Ca2+-gels at high pH. (a) Change in pH with time for 1 from the urease–urea reaction in presence of Ca2+. The black data is for no Ca2+, the red data for 0.5 equivalents of Ca2+ with respect to 1 and the blue data for 2 equivalents of Ca2+ with respect to 1. (b) Photographs showing the phase change with time when aqueous solution of the enzyme was added to the mixture of 1, urea and Ca2+: (i) immediately after addition, (ii) after 2 minutes, (iii) after 8 minutes, (iv) after 30 minutes, (v) after 1 hour, (vi) after 3 hours, (vii) after 16 hours. The white structures in the gels are air bubbles, not precipitation. (c) Variation of G′ (black), G′′ (red), tanδ (green) and pH (blue) with time for 1 involving urea–urease reaction in presence of Ca2+ ions. (d) Time dependent confocal microscopy images of 1 involving urea–urease reaction in presence of Ca2+ ions (scale bars represent 20 μm). For (a)–(d), solvent is 20/80 DMSO/H2O (v/v) and [1] = 2 mg mL−1. Initial [urease] = 0.1 mg mL−1 and [urea] = 0.01 M. For (b)–(d) [Ca2+] = 0.5 equivalents with respect to 1. | ||
The pH-dependent destruction and salt-induced reconstruction of the assemblies was studied by time sweep rheology (Fig. 3c and Fig. S14, ESI†). At low Ca2+ ion concentration, self-assembly begins (∼pH 4) immediately after the addition of H2O (containing urease) as observed from the gradual increase of G′ with time. As the pH of the medium progressively increases, G′ started to decrease and reached minimum at ∼pH 5.7 (the pKa of 1). This indicates that the primary aggregated structures are disassembling near ∼pH 5.7. However, further increase in G′ with an increase in pH confirms the reconstruction of a new type of aggregation involving the Ca2+ ions which crosslink the carboxylate groups of 1. Analysis of tanδ also confirms disassembly and rebuilding of aggregation below and above the pKa of the gelator respectively. A similar trend in the variation of G′ and tanδ was also found in presence of 2 equivalents of Ca2+ ions (Fig. S14, ESI†). At a fixed concentration of Ca2+, G′ increases as the rate of pH change is decreased.
Interestingly, at a fixed Ca2+ ion concentration, when the rate of pH change is high, a sudden decrease in G′ was noticed just after when the pH had reached a plateau at pH 9 (Fig. S14, ESI†). This was not observed when the pH change was slow. We hypothesise that when the pH change is fast, the Ca2+ binds structures reorganize again at high pH. In this case, analysis of tanδ confirms the existence of solid-like nature of the gels throughout the high pH regime.
To get more insight into the development of the microstructure of the respective gels, confocal microscopy imaging was conducted at different time intervals (Fig. 3c and Fig. S15–S17, ESI†). As soon as water is added to the DMSO solution of 1, spherulitic domains of fibres were observed. With time, these structures disappear, and new types of fibres appeared. This structural reorganization with time was more prominent when the rate of pH change was slow. Interestingly, an increase in Ca2+ ion concentration resulted in networks containing fewer spherulitic domains and instead a higher density of long fibres. Moreover, at a fixed Ca2+ ion concentration, gels obtained from slow rates of pH change were found to contain fibres which are relatively long and more interlinked than gels formed at a higher rate. We also followed the process by UV-Vis and fluorescence spectroscopy (Fig. S18–S21, ESI†). Whilst changes can be found, it is difficult to correlate the changes exactly as there are also turbidity changes throughout the process (Fig. S22, ESI†).
The final gels exhibit different mechanical properties depending on the kinetics of hydrogel formation and showed significant differences in G′ and G′′ values, as well as in gel strengths (Fig. 4a, b and Table S1, ESI†). For all the gels, G′ was considerably higher than G′′. At a fixed enzyme and urea concentration, an increase in the concentration of Ca2+ ions resulted in substantial increase in stiffness (G′) of the gels (>2 times). In the strain sweeps for all gels, both G′ and G′′ were essentially constant at low strain but deviated from linearity after a certain strain (>20% strain) indicating breaking of the gels (critical strain). At a particular Ca2+ ion concentration, the gels formed at higher rates break at higher strain and exhibited a high elastic nature (high strength) of the gels. In contrast, a decrease in gelation rate resulted in significant increase (>3 times) in G′ and G′′ of the gels. However, irrespective of formation conditions, all these gels were essentially frequency independent (Fig. S23, ESI†).
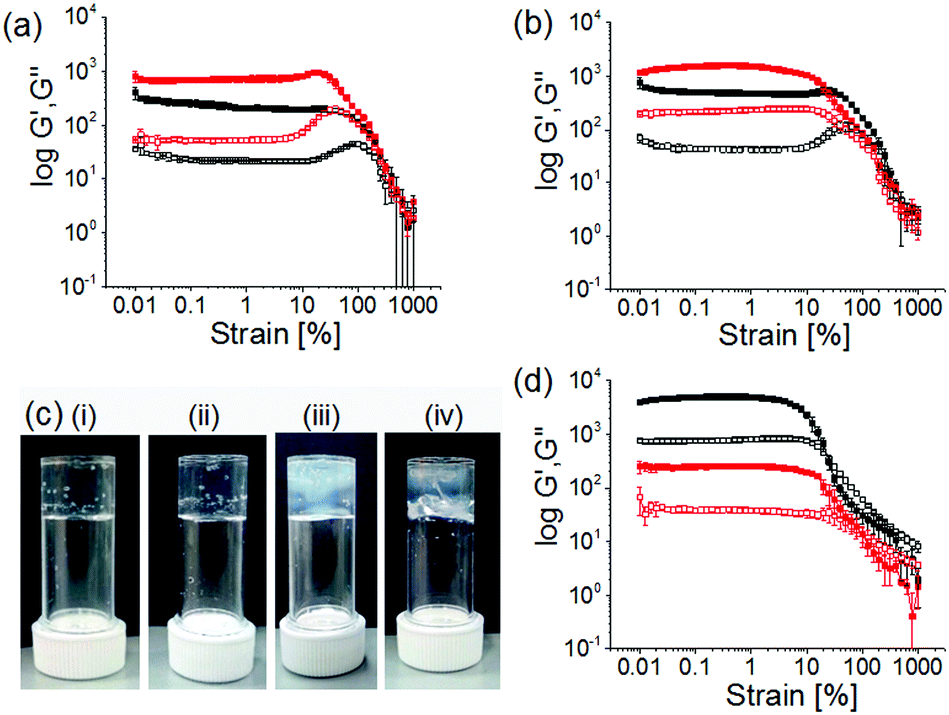 | ||
| Fig. 4 Mechanical properties of different Ca2+-triggered hydrogels. (a) and (b) strain sweep experiments of the Ca2+-triggered hydrogels ((a) 0.5 equivalents and (b) 2 equivalents with respect to 1) of 1 obtained from the dissipative pH change involving initial conditions: [urease] = 0.2 mg mL−1, [urea] = 0.02 M (black data); [urease] = 0.1 mg mL−1, [urea] = 0.01 M (red data). (c) Photographs of the different Ca2+-gels (2 molar equivalents) of 1 prepared under different conditions. Hydrogels (i) and (ii) were obtained from the dissipative pH change involving initial conditions: (i) [urease] = 0.2 mg mL−1, [urea] = 0.02 M; (ii) [urease] = 0.1 mg mL−1, [urea] = 0.01 M. Hydrogels (iii) and (iv) were prepared by external addition of 2 molar equivalents of Ca2+ ions to the solutions of 1 obtained from (iii) NaOH and (iv) enzymatic reaction involving initial conditions: [urease] = 0.1 mg mL−1, [urea] = 0.01 M. (d) Strain sweep experiments of the Ca2+-gels of 1 prepared by external addition of 2 molar equivalents of Ca2+ ions to the solutions of 1 obtained from NaOH (black data) and enzymatic reaction (red data) involving initial conditions: [urease] = 0.1 mg mL−1, [urea] = 0.01 M. For (a)–(d), [1] = 2 mg mL−1 and solvent is 20/80 DMSO/water (v/v). For (a), (b) and (d) the closed symbols represent G′, the open symbols G′′. | ||
To investigate the effectiveness of the homogeneous hydrogelation, we prepared gels by external addition of 2 molar equivalents of Ca2+ ions to the alkaline solution of 1 prepared under different conditions. The alkaline solutions were prepared in DMSO/water either by adding molar equivalents of NaOH or by performing the enzymatic reaction in absence of Ca2+ ions. Then in both cases we added Ca2+ ions on the top of the solutions and left the samples undisturbed for ∼16 hours to obtain gels. These gels showed formation of long fibers in the aggregated states (Fig. S24, ESI†). A significant difference in visual appearances was noticed as turbid inhomogeneities are prominent in these gels compared to the transparent homogeneous gels obtained through controlled pH change (Fig. 4c). In comparison to the gels formed by the dissipative process, the gels formed directly at high pH exhibits considerably higher G′, but break at a lower strain (Fig. 4d and Fig. S20, Table S2, ESI†). Evaluation of the rheological data emphasizes that gels obtained from the dissipative pH change could withstand a higher strain (20–40% strain) with higher crossover points (yield points are >450% strain where G′′ > G′), than the gels formed directly at high pH (critical strains are <10%, yield points are <100% strain). This emphasizes that this unusual gel-to-sol-to-gel approach allows access to materials that cannot be directly formed under the final conditions. Interestingly, adding a Ca2+ salt to a solution prepared by the dissipative process after the pH has increased results in a lower G′, again showing how important the process is to the final gel properties (Fig. 4d and Fig. S25, Table S2, ESI†).
Finally, our approach can be exploited to prepare an interesting case where gels are initially formed. After a pre-determined time, the return to the sol state allows flow, before the re-gelation immobilized the material once again. This is shown schematically in Fig. 5.
 | ||
| Fig. 5 Exploiting the temporal aspect of the materials. An initially formed gel (a) returns to a solution state (b). The solution state allows flow of the material from one container to a second under gravity (c), before re-gelling and immobilizing the solvent (d). Photographs taken after (a) 2 min, (b) 8 min, (c) 10 min, (d) 30 min. Note that a slight twist of the syringe barrel (with no downward force) is required at point (b) to allow air ingress and flow. Initial conditions are [1] = 2 mg mL−1, [urease] = 0.1 mg mL−1, [urea] = 0.01 M, [Ca2+] = 0.5 equivalents with respect to 1. Solvent is 20/80 DMSO/H2O (v/v). | ||
In conclusion, we have shown how a pH responsive out-of-equilibrium system can be programmed to drive an unusual gel-to-sol-to-gel transition. Unlike all other current examples of gel-based dissipative assembly, we have a system where we do not simply form a single system and then revert to the original state. Kinetic control over gelation is achieved by simple modulation of the reaction conditions that allows us to prepare homogeneous gels with improved mechanical properties. As mentioned above, there is a real need in this field for real applications to be developed. Unlike conventional dissipative assembly, where the transient formation of a gel means their use is limited to the highly specific applications where short-lived networks are needed, our gel-to-sol-to-gel transition allows us to prepare gels with properties that cannot be directly accessed for example. This makes our approach analogous to annealing.
S. P. thanks the Royal Society and SERB of India for a Newton International Fellowship. D. A. thanks the EPSRC for a Fellowship (EP/L021978/1).
Conflicts of interest
There are no conflicts to declare.Notes and references
- P. Terech and R. G. Weiss, Chem. Rev., 1997, 97, 3133–3160 CrossRef CAS PubMed.
- L. A. Estroff and A. D. Hamilton, Chem. Rev., 2004, 104, 1201–1218 CrossRef CAS PubMed.
- S. S. Babu, V. K. Praveen and A. Ajayaghosh, Chem. Rev., 2014, 114, 1973–2129 CrossRef CAS PubMed.
- X. Du, J. Zhou, J. Shi and B. Xu, Chem. Rev., 2015, 115, 13165–13307 CrossRef CAS PubMed.
- B. Rieß, C. Wanzke, M. Tena-Solsona, R. K. Grötsch, C. Maity and J. Boekhoven, Soft Matter, 2018, 14, 4852–4859 RSC.
- S. Panettieri and R. V. Ulijn, Curr. Opin. Struct. Biol., 2018, 51, 9–18 CrossRef CAS PubMed.
- B. Rieß and J. Boekhoven, ChemNanoMat, 2018, 4, 710–719 CrossRef.
- S. De and R. Klajn, Adv. Mater., 2018, 30, 1706750 CrossRef PubMed.
- M. Tena-Solsona and J. Boekhoven, Isr. J. Chem., 2019, 59, 1–9 CrossRef.
- S. A. P. van Rossum, M. Tena-Solsona, J. H. van Esch, R. Eelkema and J. Boekhoven, Chem. Soc. Rev., 2017, 46, 5519–5535 RSC.
- C. G. Pappas, I. R. Sasselli and R. V. Ulijn, Angew. Chem., Int. Ed., 2015, 54, 8119–8123 CrossRef CAS PubMed.
- S. Bal, K. Das, S. Ahmed and D. Das, Angew. Chem., Int. Ed., 2019, 58, 244–247 CrossRef CAS PubMed.
- T. Heuser, E. Weyandt and A. Walther, Angew. Chem., Int. Ed., 2015, 54, 13258–13262 CrossRef CAS PubMed.
- J. P. Wojciechowski, A. D. Martin and P. Thordarson, J. Am. Chem. Soc., 2018, 140, 2869–2874 CrossRef CAS PubMed.
- S. M. Morrow, I. Colomer and S. P. Fletcher, Nat. Commun., 2019, 10, 1011 CrossRef PubMed.
- J. Boekhoven, A. M. Brizard, K. N. K. Kowlgi, G. J. M. Koper, R. Eelkema and J. H. van Esch, Angew. Chem., Int. Ed., 2010, 49, 4825–4828 CrossRef CAS PubMed.
- J. Boekhoven, W. E. Hendriksen, G. J. M. Koper, R. Eelkema and J. H. van Esch, Science, 2015, 349, 1075 CrossRef CAS PubMed.
- M. Tena-Solsona, B. Rieß, R. K. Grötsch, F. C. Löhrer, C. Wanzke, B. Käsdorf, A. R. Bausch, P. Müller-Buschbaum, O. Lieleg and J. Boekhoven, Nat. Commun., 2017, 8, 15895 CrossRef CAS PubMed.
- A. Jain, S. Dhiman, A. Dhayani, P. K. Vemula and S. J. George, Nat. Commun., 2019, 10, 450 CrossRef PubMed.
- C. A. Angulo-Pachón and J. F. Miravet, Chem. Commun., 2016, 52, 5398–5401 RSC.
- B. A. Grzybowski, K. Fitzner, J. Paczesny and S. Granick, Chem. Soc. Rev., 2017, 46, 5647–5678 RSC.
- C. Colquhoun, E. R. Draper, R. Schweins, M. Marcello, D. Vadukul, L. C. Serpell and D. J. Adams, Soft Matter, 2017, 13, 1914–1919 RSC.
- L. Chen, K. Morris, A. Laybourn, D. Elias, M. R. Hicks, A. Rodger, L. Serpell and D. J. Adams, Langmuir, 2010, 26, 5232–5242 CrossRef CAS PubMed.
- E. Jee, T. Bánsági Jr, A. F. Taylor and J. A. Pojman, Angew. Chem., Int. Ed., 2016, 55, 2127–2131 CrossRef CAS PubMed.
- S. Panja, C. Patterson and D. J. Adams, Macromol. Rapid Commun., 2019, 0, 1900251 CrossRef PubMed.
- S. Panja and D. J. Adams, Chem. Commun., 2019, 55, 47–50 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9cc05154f |
| This journal is © The Royal Society of Chemistry 2019 |