
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFully independent photochemical reactivity in one molecule†
David E.
Marschner‡
ab,
Christoph O.
Franck‡
ac,
Doris
Abt
a,
Hatice
Mutlu
 *ad and
Christopher
Barner-Kowollik
*ab
*ad and
Christopher
Barner-Kowollik
*ab
aMacromolecular Architectures, Institute for Chemical Technology and Polymer Chemistry, Karlsruhe Institute of Technology (KIT), Engesserstr. 18, 76131 Karlsruhe, Germany. E-mail: christopher.barner-kowollik@kit.edu
bSchool of Chemistry, Physics and Mechanical Engineering, Queensland University of Technology (QUT), 2 George Street, Brisbane, QLD 4000, Australia. E-mail: christopher.barnerkowollik@qut.edu.au
cDepartment of Chemical Engineering and Biotechnology, University of Cambridge Philippa Fawcett Drive, Cambridge, CB3 0AS, UK
dSoft Matter Synthesis Laboratory, Institute for Biological Interfaces, Karlsruhe Institute of Technology (KIT), Hermann-von-Helmholtz-Platz 1, 76344 Karlsruhe, Germany. E-mail: hatice.mutlu@kit.edu
First published on 22nd July 2019
Abstract
We introduce a chemically λ-orthogonal bichromophore triggered simply by different colours of light, enabling two different photochemical reactions in one molecule. Uniquely, the short wavelength (λ = 314 nm) does not trigger the red-shifted reaction system (λ = 416 nm), opening possibilities for the light controlled gating of specific molecular sites independent of wavelength.
Beyond fundamental scientific interest, a key quest in modern synthetic chemistry is to achieve spatiotemporal control over two (or more) different reactive moieties in a one-pot reaction mixture.1,2 Indeed, such control can be readily achieved by adopting orthogonal light-harvesting reactions.2 However, the use of such reactions is often challenging due to wavelength-dependent selectivity issues, since many reactions utilise light in the same range of wavelengths.3 For an independent reaction process, it is necessary to separate the absorption bands of the employed chromophores and therefore their respective triggers, e.g. through red-shifting.2 Only a few examples of wavelength-orthogonal (λ-orthogonal) reaction systems have been reported so far, many of which are beset with limitations such as a specific order that needs to be followed in respect to reaction sequence.4 Often, incomplete orthogonality is observed since the visible light reactive component is also triggered when using UV light. Thus, the reaction is only orthogonal when irradiation takes place starting with the longest wavelength and then proceeding to the shorter one. To overcome this sequence dependent procedure, it is possible to use reactions that are either deactivated or unreactive in the visible light range. Indeed, a pathway to complete λ-orthogonality is provided by the utilisation of reversible photochemistry, for example through the use of styrylpyrene, as it exclusively dimerises when irradiated with visible light and not under UV-A irradiation.5,6 In fact, it was recently reported that styrylpyrene can be combined with other light-harvesting groups such as anthracene to enable an additional second dimerisation reaction.7,8 An alternative chromophore that can undergo a photoreaction under UV-A light while remaining unreactive in the visible light range is o-methyl benzaldehyde (o-MBA), which generates an o-quinodimethane species under excitation with light that readily reacts with maleimides.9 Compared to anthracene, o-MBA has the advantage that it can react with various molecules featuring double bonds (e.g. maleimides, fumarates), while anthracene is able to undergo dimerisation with itself only. In combination with both a reversible as well as visible light system, this could pave a way for novel λ-orthogonal systems.
The motivation underpinning the current study is to combine several light-harvesting chromophores into one molecule that can undergo site specific cycloaddition reactions at different wavelengths in a λ-orthogonal fashion. By choosing different linkage molecules, e.g. star linkers, this idea can be expanded from a system with two functional groups to multiple reactions sites – a polychromophoric molecule. To demonstrate our concept, establish a platform technology and enable in-depth analytical characterisation, a bi-linker was selected as concept molecule. o-MBA was one of the light-responsive moieties of choice, which can be triggered with UV-A light to react with preferably electron-deficient double bonds, such as maleimides, yet can also self-dimerise. Nevertheless, in previous studies we evidenced that o-MBA preferentially reacts in an efficient manner with activated dienes, such as maleimides, than undergoing self-dimerisation.10,11 In addition, o-MBA (310–350 nm) has shown to be suitable for sequence-dependent λ-orthogonal reactions in combination with tetrazoles (270–310 nm), where the higher wavelength component is fully triggered first.10 Recently, we demonstrated that o-MBA and tetrazoles can be used in a sequence-independent λ-orthogonal fashion, however, only through control of the respective number of photons and solvents used during irradiation.12 The second chromophore is a cinnamic acid derivative, which can undergo a reversible [2+2] photo-cycloaddition. To achieve a sufficient separation of the absorption bands – and thus their triggering wavelengths – for the two light-harvesting groups, functionalisation of the cinnamic acid with a pyrene unit is preferential as it is known to red-shift double bonds that can subsequently undergo a [2+2] cycloaddition within the visible light regime (e.g. styrylpyrene, pyrene aryl tetrazole).6,13,14 With such a bichromophoric molecule, it is anticipated that two different pathways completely independent of each other may result in the same product as depicted in Fig. 1.
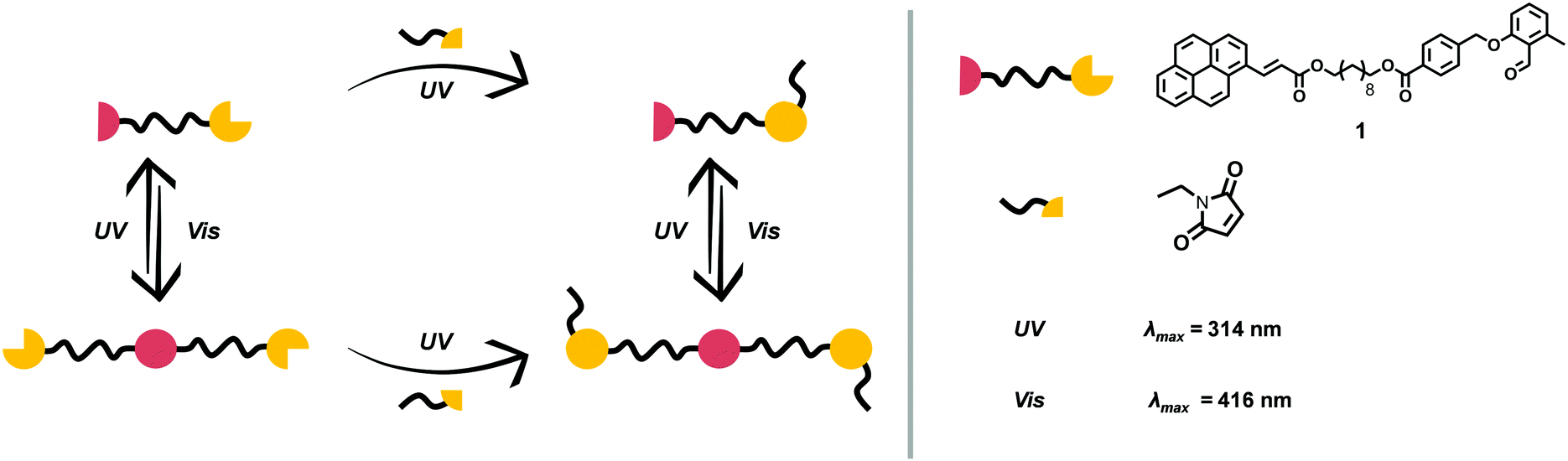 | ||
| Fig. 1 Schematic overview showing the investigated bichromophore with its pyrenyl cinnamic acid (PCA) and ortho-methylbenzaldehyde (o-MBA) end groups as well as the anticipated orthogonal synthesis platform possible upon irradiation with light. | ||
To determine the reactivity of the cinnamic acid double bond, a proof-of-concept molecule was synthesised. Specifically, a pyrenyl cinnamic acid (PCA) derivative (2, refer to Fig. 3) featuring a butanol chain instead of being linked to o-MBA. 2 was used to study the dimerisation as well as the dissociation behaviour of the double bond without the possibility of triggering the o-MBA moiety, especially when irradiating the molecule with UV-A light. As expected, functionalisation of cinnamic acid with pyrene introduced a red-shift of ca. 100 nm to λmax = 370 nm of the resulting compound when compared to cinnamic acid: λmax = 272 nm, which can be explained by the extension of the conjugated system.6,14,15 Exclusively under irradiation with visible light (λmax = 416 nm) 2 undergoes dimerisation, but also isomerisation, evidenced by NMR (refer to ESI,† Fig. S1–S3). In addition, the UV/Vis spectra indicate the formation of the targeted photoproduct (Fig. 2). When 2 undergoes dimerisation, the extended conjugation system is reduced, and the molecule converts to an unconjugated spectrum featuring two absorption bands at λ = 333 and 348 nm, indicating a successful photoreaction.16 According to absorbance measurements, maximum conversion (79% photoproduct, calculated from 1H NMR spectra via integration of distinct resonances for the different species) was reached after 60 min (refer to ESI,† Fig. S4A).
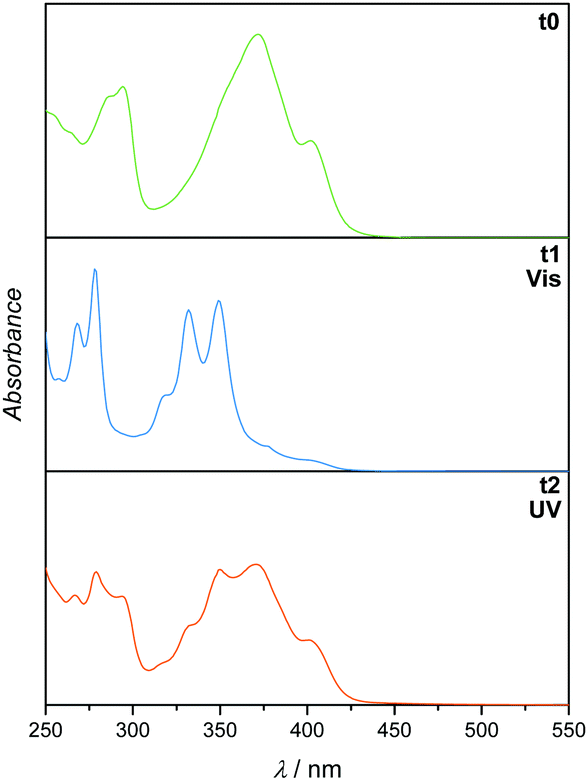 | ||
| Fig. 2 UV/Vis spectra (recorded in DCM) of 2 before irradiation (t0), after irradiation with visible light (t1) and subsequent irradiation with UV light (t2). | ||
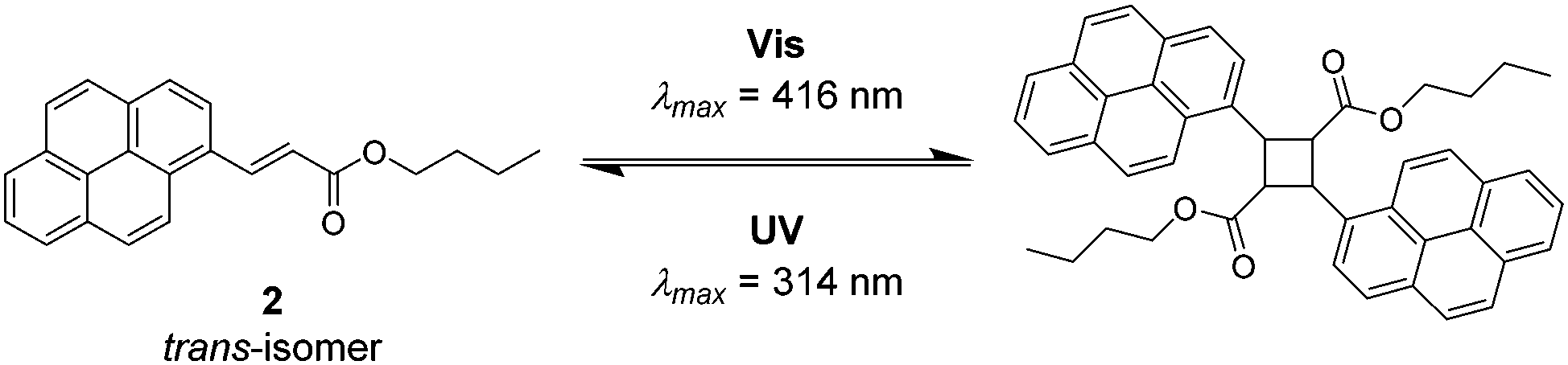 | ||
| Fig. 3 Reversible [2+2] cycloaddition of pyrenyl cinnamic acid 2 under irradiation with visible (λmax = 416 nm) and irradiation with UV-A light (λmax = 314 nm). Irradiation with UV-A light introduces not only the cycloreversion, yet also the isomerisation of 2. | ||
In theory, [2+2] cycloadditions are reversible and can be reverted to their starting materials once they are irradiated with a wavelength that is shorter than the one required for the respective dimerisation reaction.3 In order to investigate the cycloreversion, the 2-dimer was irradiated with UV-A light (λmax = 314 nm). Consequently, the reaction mixtures were characterised in-depth by utilising NMR spectroscopy which showed almost complete reversion of the dimer (3% remaining dimer, calculated from 1H NMR spectra) to the starting trans-isomer of 2 that partly underwent photoisomerisation to its cis-structure (ESI,† Fig. S1 and S2). In a similar fashion to the dimerisation reaction, dissociation was observed after 60 min (according to the absorbance measurements, refer to the ESI,† Fig. S4B). The remaining traces of dimeric structures can also be observed in the UV/Vis spectra since the trace from t1 did not revert to t0, but rather displays a composite of the two traces. However, because the absorbance spectra of all compounds (trans- and cis-2, as well as the photoproducts) are overlapping, they cannot be used to determine the exact conversion. To ensure that the molecules are not affected during irradiation, the samples were analysed via GC-MS (ESI,† Fig. S5–S7), though no fragments with smaller mass values could be detected confirming that no photodamage took place.
Next, the o-MBA functionality in bichromophore 1 was investigated. Irradiation of 1 with UV-A light (λmax = 314 nm) should introduce a [4+2] cycloaddition of o-MBA with N-ethyl maleimide (Et-Mal). Indeed, subsequent analysis via NMR spectroscopy showed complete consumption of both o-MBA's aldehyde and methyl resonances (refer to ESI,† Fig. S7).
With the irreversible ligation site of 1 fully functional, the visible light triggered reversible PCA was subsequently investigated. When 1 was irradiated with λmax = 416 nm, the double bond dimerised forming a cyclobutane structure that was reversible under UV-A irradiation (λmax = 314 nm). The reaction was analysed via1H NMR (ESI,† Fig. S10) as well as UV/Vis spectroscopy (ESI,† Fig. S9) and the results were completely analogous to the small molecule study of 2. Comparable to the test molecule 2, 1H NMR showed new magnetic resonances for the dimerisation reaction that almost completely disappeared during dissociation. UV/Vis spectroscopy depicted the decrease of the broad absorption band representing the fully conjugated system (λmax = 371 nm) of the trans-isomer. Subsequent irradiation with UV-A light did not convert the spectrum back to its starting pattern but a mixture of monomeric and dimeric structures, which is similar to that of the small molecule study (2).
Having successfully demonstrated both the irreversible [4+2] cycloaddition of o-MBA and the reversible [2+2] cycloaddition of PCA along with their independence from each other, the complete synthesis platform with subsequent orthogonal and selective absorption was carried out (refer to Fig. 1). As expected from the small molecule study, the reaction system can be irradiated with UV light triggering the o-MBA unit to undergo a reaction with Et-Mal. 1H NMR clearly shows the consumption of the o-MBA (refer to ESI,† Fig. S10). However, the double bond underwent isomerisation from the trans- to the cis-structure, alongside small amounts of dimerisation, which can be detected via NMR measurements, and small amounts of the previously discussed absorption pattern during UV/Vis spectroscopy (Fig. 4). A plausible reason for the traces of dimer molecules is the broad emission spectrum of the utilised UV lamps that also feature small emissions up to wavelengths of approximately 400 nm (refer to the ESI,† Fig. S24). Subsequent irradiation of the same mixture with visible light (λmax = 416 nm) introduced dimerisation of the double bond (Fig. 4), successfully proving the reaction pathway utilising UV and succeeding visible light irradiation of the reaction system shown in Fig. 1.
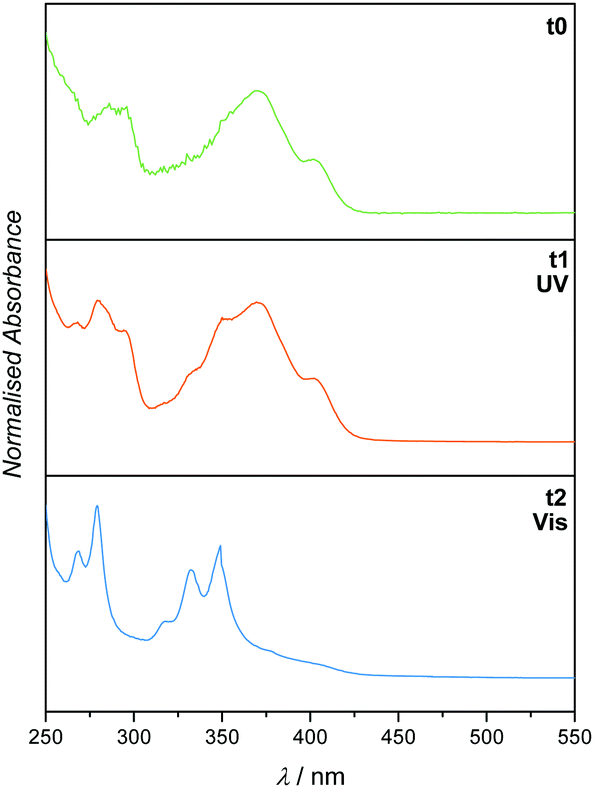 | ||
| Fig. 4 UV/Vis spectra (recorded in DCM) of 1 before irradiation (t0), after irradiation with UV light (t1) and subsequent irradiation with visible light (t2). Irradiation took place in the presence of Et-Mal. | ||
To explore the complementary route (depicted in Fig. 1), the sequence of irradiation was reversed by first treating the starting material 1 with visible light. The result indicated the successful dimerisation of the double bond, while both the aldehyde and methyl functionalities of o-MBA remained intact. Subsequent irradiation with UV light, however, induced not only the irreversible cyclisation reaction of o-MBA, but also the dissociation of the cyclised product generated from the previous step due to the reversible nature of the pyrenyl cinnamic acid system (refer to Fig. 5). Isomerisation of the double bonds also occurred during the UV treatment. Therefore, the complete wavelength-orthogonality only applied to the first step, where the uniquely designed compound 1 was involved. Although the UV treatment in the second step could not trigger the reaction selectively, it produced the same intermediate product as the previous route, and the re-exposure of it to visible light can again trigger the dimerisation reaction, and generate the desired final product (refer to Fig. 6).
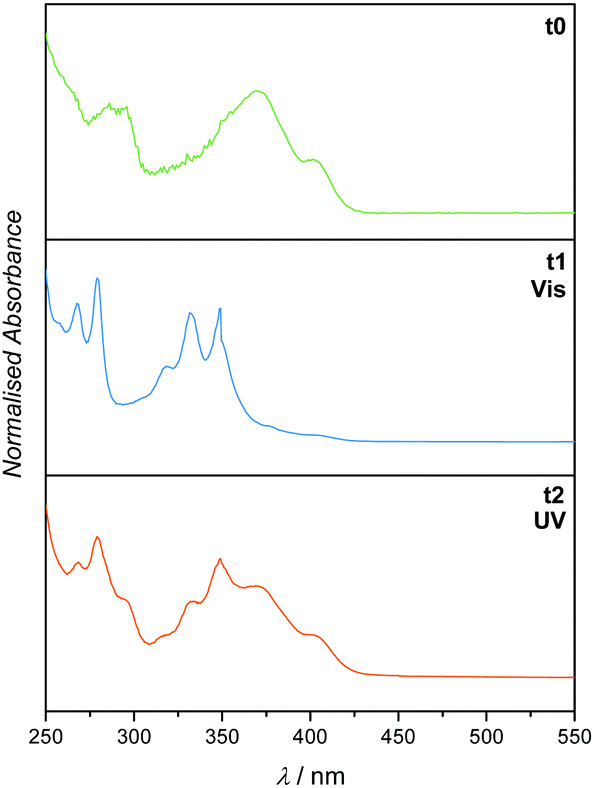 | ||
| Fig. 5 UV/Vis spectra (recorded in DCM) of 1 before irradiation (t0), after irradiation with visible light (t1) and subsequent irradiation with UV light (t2). Irradiation took place in the presence of Et-Mal. | ||
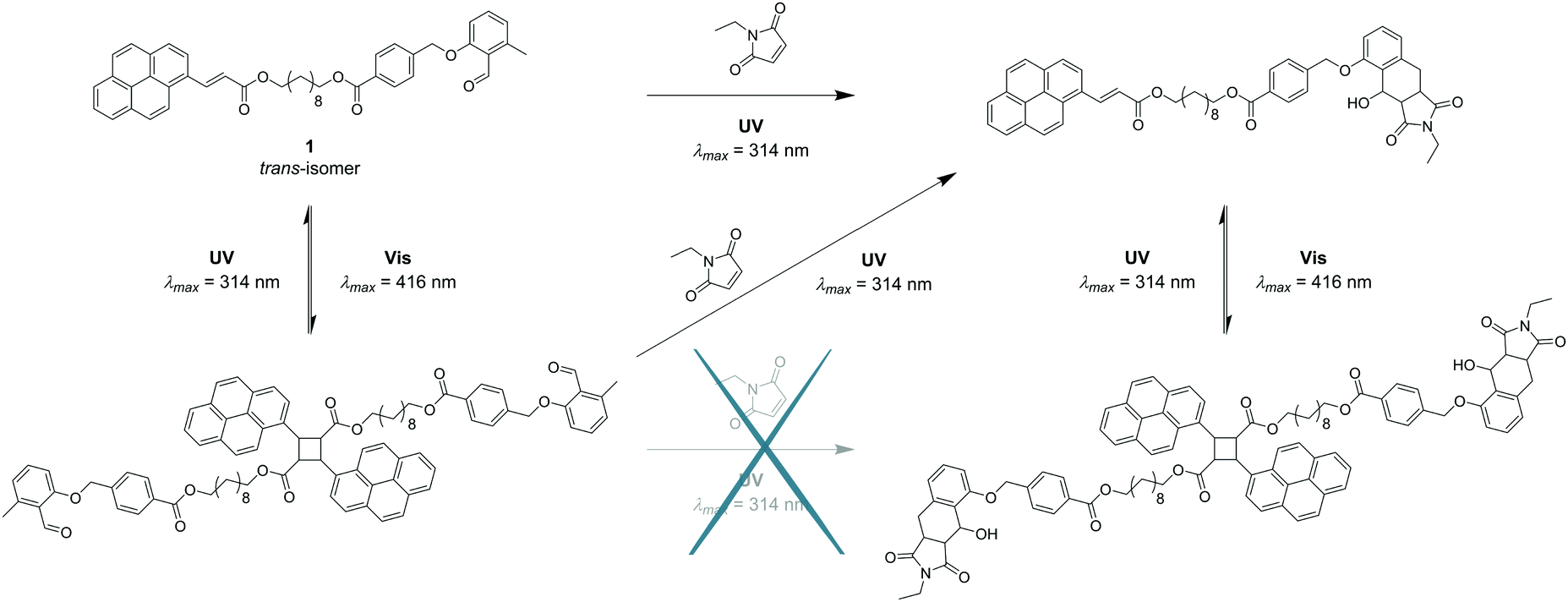 | ||
| Fig. 6 Reaction scheme of 1 showing the anticipated orthogonal synthesis platform under irradiation with light as well as the pathways that were established via analysis with NMR and UV/Vis spectroscopy. The bichromophore 1 can undergo a first λ-orthogonal reaction by irradiation with either visible or UV light, while the subsequent inversion of the irradiation sequence results in the same final product. | ||
All described molecules proved to be bench stable for over six months at ambient conditions, suggesting a high resistance against degradation, non-triggered cycloaddition or unwanted side reactions. In addition, initial tests that replaced Et-Mal with a polyethylene glycol featuring maleimide as an end group showed promising results (refer to ESI,† Chapter S1.2), suggesting that this system could also be applicable in polymer chemistry and materials science, which will be explored in future research.
In summary, we introduce a light-harvesting system in which two functional units are orthogonal to each other within the same molecule. These groups can undergo two different, side-specific photoreactions independently of each other within one molecule, exclusively by the choice of different wavelengths (UV-A and visible light). It was observed that in subsequent steps alternating UV and visible light irradiation, the same end product can be obtained. The molecule features highly interesting properties including bench stability, which enables a high sample portability. In future research, the photoreactive groups may be exchanged allowing the system to be finetuned to different applications or to introduce subsequent λ-orthogonal reaction steps. In addition, the bi-linker could be replaced with a linker featuring multiple light-harvesting groups for the synthesis of a probe that can be used as a portable and stable wavelength marker for various light regimes. The expansion of the shown concept from bi- to polychromophores opens exciting possibilities not only as wavelength markers, yet also as dynamic and reprogrammable materials. Especially through incorporation of more than one photo reversible light-harvesting group, such a polychromophore linker could be used to tailor materials to different applications and further reprogramming for specific needs. By irradiating these functional materials with different wavelengths, another part of the linker is triggered to further crosslink or – in the case of reversible photoreactions – de-crosslink the materials making them stiffer, softer or changing other physical properties such as colour. Most importantly, all these applications are combined in a completely λ-orthogonal single molecule that also features a long lifetime without any degradation processes taking place.
C. B.-K. acknowledges funding from the Karlsruhe Institute of Technology (KIT), the Australian Research Council (ARC) via a Laureate Fellowship underpinning his photochemical research program and the German Research Council (DFG). C. B.-K. acknowledges additional support under Germany's Excellence Strategy via the Excellence Cluster 3D Matter Made to Order (EXC-2082-390761711).
Conflicts of interest
There are no conflicts to declare.Notes and references
- A. Blanc and C. G. Bochet, J. Org. Chem., 2002, 67, 5567–5577 CrossRef CAS PubMed.
- H. Frisch, D. E. Marschner, A. S. Goldmann and C. Barner-Kowollik, Angew. Chem., Int. Ed., 2018, 57, 2036–2045 CrossRef CAS PubMed.
- G. Kaur, P. Johnston and K. Saito, Polym. Chem., 2014, 5, 2171–2186 RSC.
- M. J. Hansen, W. A. Velema, M. M. Lerch, W. Szymanski and B. L. Feringa, Chem. Soc. Rev., 2015, 44, 3358–3377 RSC.
- V. X. Truong, F. Li, F. Ercole and J. S. Forsythe, ACS Macro Lett., 2018, 7, 464–469 CrossRef CAS.
- D. E. Marschner, H. Frisch, J. T. Offenloch, B. T. Tuten, C. R. Becer, A. Walther, A. S. Goldmann, P. Tzvetkova and C. Barner-Kowollik, Macromolecules, 2018, 51, 3802–3807 CrossRef CAS.
- H. Frisch, F. R. Bloesser and C. Barner-Kowollik, Angew. Chem., Int. Ed., 2019, 58, 3604–3609 CrossRef CAS PubMed.
- S. Bialas, L. Michalek, D. E. Marschner, T. Krappitz, M. Wegener, J. Blinco, E. Blasco, H. Frisch and C. Barner-Kowollik, Adv. Mater., 2019, 1807288 CrossRef PubMed.
- T. Gruendling, K. K. Oehlenschlaeger, E. Frick, M. Glassner, C. Schmid and C. Barner-Kowollik, Macromol. Rapid Commun., 2011, 32, 807–812 CrossRef CAS PubMed.
- K. Hiltebrandt, M. Kaupp, E. Molle, J. P. Menzel, J. P. Blinco and C. Barner-Kowollik, Chem. Commun., 2016, 52, 9426–9429 RSC.
- T. Gegenhuber, L. De Keer, A. S. Goldmann, P. H. M. Van Steenberge, J. O. Mueller, O. Reyniers, J. P. Menzel, D. R. D’hooge and C. Barner-Kowollik, Macromolecules, 2017, 50, 6451–6467 CrossRef CAS.
- J. P. Menzel, F. Feist, B. Tuten, T. Weil, J. P. Blinco and C. Barner-Kowollik, Angew. Chem., Int. Ed., 2019, 58, 7470–7474 CrossRef CAS PubMed.
- N. P. Kovalenko, A. T. Abdukadyrov, V. I. Gerko and M. V. Alfimov, J. Photochem., 1980, 12, 59–65 CrossRef CAS.
- P. Lederhose, K. N. R. Wüst, C. Barner-Kowollik and J. P. Blinco, Chem. Commun., 2016, 52, 5928–5931 RSC.
- E. Havinga and R. J. F. Nivard, Recl. Trav. Chim. Pays-Bas, 1948, 67, 846–854 CrossRef CAS.
- T. Doi, H. Kawai, K. Murayama, H. Kashida and H. Asanuma, Chem. – Eur. J., 2016, 22, 10533–10538 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: In-depth analysis including NMR, UV/Vis and GC-MS spectra providing further proof of the introduced concept. See DOI: 10.1039/c9cc04781f |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2019 |