
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Graphene–aramid nanocomposite fibres via superacid co-processing†
Aled D.
Roberts
 ab,
Paul
Kelly
a,
Jennifer
Bain
ab,
John J.
Morrison
c,
Ian
Wimpenny
b,
Mike
Barrow
d,
Robert T.
Woodward
e,
Matthieu
Gresil
b,
Christopher
Blanford
b,
Sam
Hay
a,
Jonny J.
Blaker
b,
Steve G.
Yeates
*c and
Nigel S.
Scrutton
*a
ab,
Paul
Kelly
a,
Jennifer
Bain
ab,
John J.
Morrison
c,
Ian
Wimpenny
b,
Mike
Barrow
d,
Robert T.
Woodward
e,
Matthieu
Gresil
b,
Christopher
Blanford
b,
Sam
Hay
a,
Jonny J.
Blaker
b,
Steve G.
Yeates
*c and
Nigel S.
Scrutton
*a
aManchester Institute of Biotechnology, The University of Manchester, M1 7DN, UK. E-mail: Nigel.Scrutton@manchester.ac.uk
bSchool of Materials, The University of Manchester, Manchester, M1 2PG, UK
cSchool of Chemistry, The University of Manchester, Oxford Road, Manchester, M13 9PL, UK. E-mail: Stephen.Yeates@manchester.ac.uk
dAnton Paar Ltd. Unit F, The Courtyard St. Albans, AL4 0LA, UK
eDep. of Chemical Engineering, Imperial College London, London, SW7 2AZ, UK
First published on 11th September 2019
Abstract
The development of graphene–polymer nanocomposite materials has been hindered by issues such as poor colloidal stability of graphene in liquid media, weak interactions between graphene and the host polymers as well as the lack of scalable and economical graphene synthesis routes. Chlorosulfonic acid (CSA) can spontaneously disperse graphene without the need for mechanical agitation, chemical functionalisation or surfactant stabilisation,1 however is incompatible with most polymers and organic materials. Here, we demonstrate how poly(p-phenylene terephthalamide) (PPTA) – the polymer which constitutes Kevlar – can be co-processed with graphene in CSA and wet-spun into nanocomposite fibres with minimal aggregation of graphene.
A single layer of pristine graphene has an intrinsic strength of ca. 130 GPa and a Young's modulus (stiffness) of ca. 1.0 TPa, greatly exceeding the mechanical properties of any known bulk material.2 Despite these outstanding properties, attempts to commercialise graphene-based materials have been frustrated by issues such as its poor colloidal stability in most liquid phases, the lack of economical and scalable graphene synthesis routes, as well as poor interactions between graphene and polymer matrixes.3,4 Nevertheless, the incorporation of even small quantities of graphene into host polymers has been shown to significantly enhance their mechanical, electrical and thermal properties.4–6
Aromatic polyamides (aramids) are a class of synthetic polymers which, when spun from liquid crystalline dopes under carefully controlled conditions, can form paracrystalline fibres with exceptionally high strength, stiffness and toughness.7–10 For these reasons, aramid fibres such as Kevlar, Twaron and Nomex are employed for a range of high-performance applications where a high strength-to-weight ratio is needed, such as ballistic armour, car-tyre reinforcements and aerospace composites.10 The incorporation of graphene into aramid fibres may further enhance their mechanical properties, as well as introduce electrical conductivity, as has been shown for a myriad of other graphene–polymer nanocomposites.4,5 The relatively linear nature of aramid polymers, coupled with their high degree of aromaticity, could also stabilise graphene sheets through π–π interactions, as well as effectively transfer mechanical stress from the polymer matrix to the reinforcing graphene.11–14
Graphene can be produced through a number of different routes, which can be classified as either top-down or bottom-up approaches.4,5 Of these, the direct exfoliation of graphite by the superacid chlorosulfonic acid (CSA) is considered to be one of the most effective methods for its scalable and economical production.1,4,15,16 By protonating and spontaneously exfoliating graphite, CSA yields colloidal liquid crystal dispersions of high-purity, single-layer graphene.1 Moreover, this is achieved without the need for mechanical agitation, chemical functionalisation or surfactant stabilisation – which can compromise graphene's outstanding physical properties.4,5 A major drawback of this technique is that CSA has poor compatibility with most conventional materials, reacting violently with water, alcohols, metals, and organic materials including many polymers and solvents.17
Aramids are one of the few organic materials that can be dissolved by, yet resist decomposition from, powerful acids such as CSA – and are typically spun into fibres from anhydrous fuming sulfuric acid solutions at elevated temperatures.9 In this communication, we show how PPTA–graphene composite fibres can be produced via co-processing with CSA. Wide-angle X-ray diffraction (WAXD) and polarised light microscopy indicated minimal aggregation of 2 μm graphene sheets within the fibres, even at a relatively high loading of 10% w/w. Evolution of gases (e.g., H2O, HCl vapour) at the point of fibre formation (due to contact of CSA with the water coagulation bath) resulted in porous fibres with large cavities and a specific surface area of ca. 16 m2 g−1. This porosity was likely detrimental to the mechanical and electrical conductivity properties of the fibres, meaning further development of the fibre spinning technique and processing conditions, such as post-spin drawing and heat treatment, would likely be required to attain high-strength, non-porous fibres analogous to commercial aramid fibres. Improved spinning conditions (e.g., employing dry-jet wet spinning) would also be needed to increase the degree of molecular and crystallite orientation to further improve mechanical properties, as seen in commercial aramid fibres.9,10,18,19
In this work, PPTA was synthesised through the low-temperature anhydrous polycondensation of benzene-1,4-dicarbonyl dichloride and benzene-1,4-diamine (details in ESI†) (Fig. 1).20 Commercial aramid fibres were not reprocessed for spinning due to the presence of contaminants (e.g., finishing oils) and since their precise composition is not disclosed by manufacturers (e.g., may contain up to 15 mol% non-aromatic linkages).9,10 Since fibre strength is proportional to average polymer molecular weight (Mave),18 the synthesis was first subject to several rounds of optimisation to maximise Mave. Due to the poor solubility of PPTA in most solvents, common techniques such as gel permeation chromatography (GPC) could not be employed to determine Mave. Instead, capillary viscometry was employed to compare the dynamic viscosity (η) of the batches under analogous conditions, where a higher η implied a higher Mave (Table S1, ESI†). Multiple dilute solution viscometry measurements were employed to obtain approximate Mave values for the optimised batch (abbreviated B7-PPTA) as well as commercial Kevlar, by employing both the Huggins and Kraemer methods followed by application of the Mark–Houwink equation:
| [η] = KMαave |
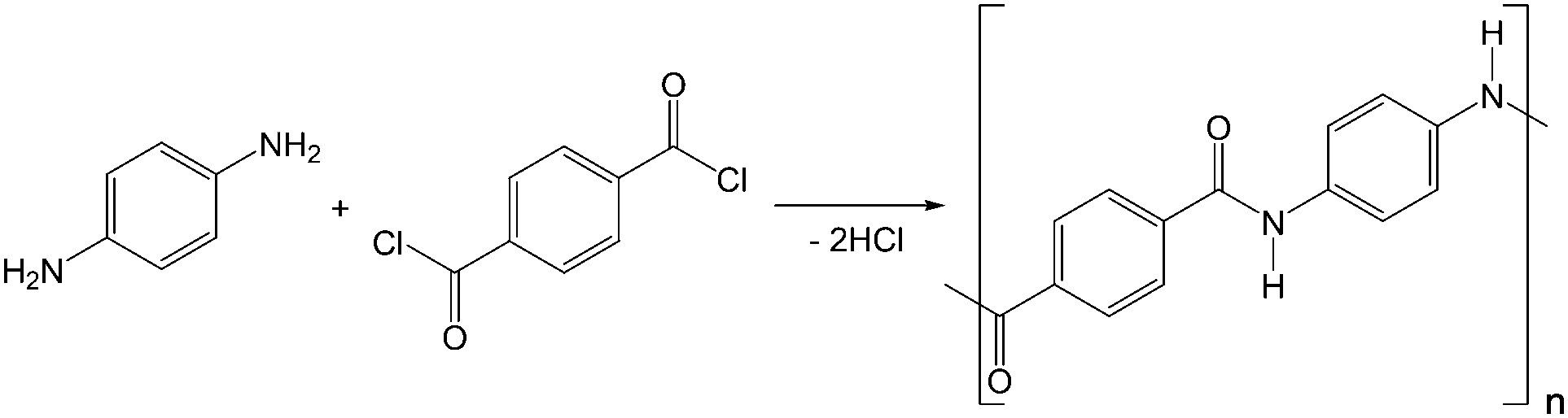 | ||
| Fig. 1 Reaction scheme showing synthesis of PPTA from TPC and PPD. | ||
To produce PPTA–graphene spinning dopes, two grades of graphite nanoplatelets (GNPs) – M25 and C750 – were first dispersed in CSA before addition and dissolution of B7-PPTA (details in ESI†). M25 GNPs have a stated average platelet diameter of ca. 25 μm whilst C750 GNPs have an average platelet diameter of ca. 2 μm.22 The resultant PPTA spinning dopes (12% w/w) had graphene contents between 0.1–10% by mass relative to the PPTA (Fig. S1, ESI†). The solutions were then subject to wet spinning into a water coagulation bath using a custom-made spinning rig (Fig. S2, ESI†). After washing and drying, the B7-PPTA fibres without graphene had a pale yellow/brown colouration, which darkened through green into black as the graphene content increased (Fig. 2a). Fourier transform infrared spectroscopy (FTIR) was performed on all samples, showing an almost identical signature to commercial Kevlar (Fig. S3, ESI†). Scanning electron microscopy (SEM) images of the fibres revealed a relatively rough surface compared to commercial Kevlar (Fig. 2b). The thickness of the fibres varied between ca. 45–120 μm in diameter, significantly larger than commercial Kevlar which are ca. 11 μm in diameter. Smaller diameter fibres with greater uniformity could likely be obtained with a commercial wet-spinning rig. Polarised light microscopy revealed the B7-PPTA fibres to be paracrystalline, as observed by the birefringence effect (i.e. rotation of polarised light by crystalline domains) (Fig. 2c). This effect was exploited to visualise the dispersion of graphene throughout the fibres: as graphene content increased, the intensity of light passing through the fibres decreased – highlighting any regions of aggregated graphene as substantially darker areas. It can be seen from Fig. 2c that the B7-PPTA fibres containing M25 graphene have these darker areas, suggesting graphene aggregation, whereas the fibres with C750 darken relatively uniformly suggesting good dispersion.
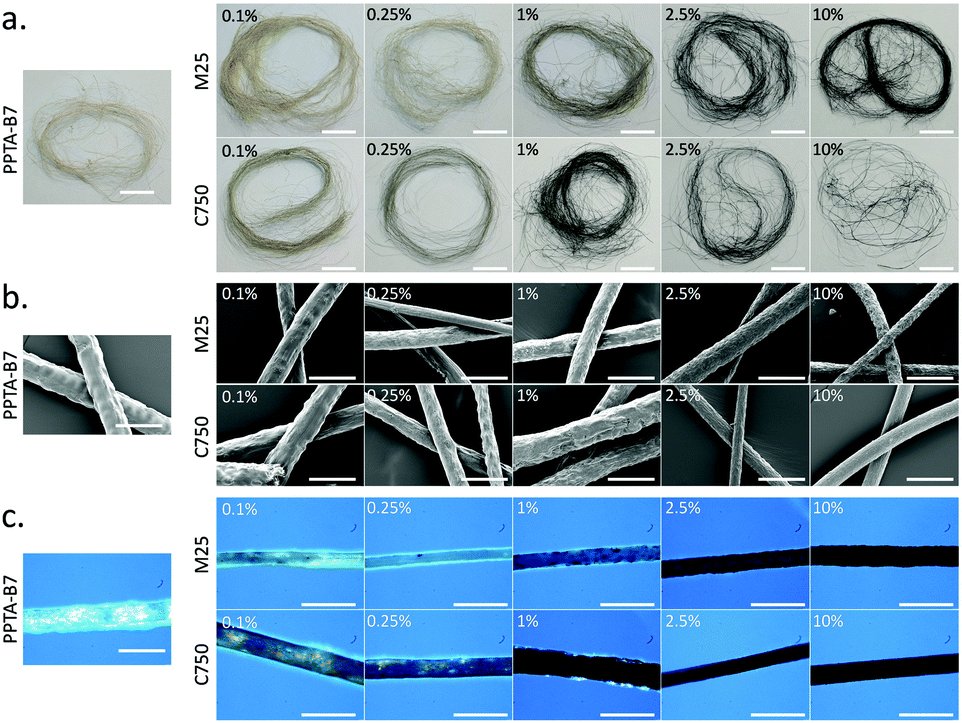 | ||
| Fig. 2 (a) Visible-light images (scale bars ca. 2 cm), (b) SEM images (scale bars 200 μm) and (c) cross-polarised light microscopy images (scale bars 200 μm) of wet-spun B7-PPTA fibres with increasing loading (0.1–10% by mass) of C750 and M25 graphene. | ||
WAXD was employed to further probe the dispersion of the graphene as well as the crystallinity of the fibres (Fig. 3). Without graphene, the diffraction peaks of the B7-PPTA fibres matched those of commercial Kevlar,8 although were notably broader suggesting smaller crystallites and hence a less ordered paracrystalline structure.10 Commercial aramid fibres also have highly anisotropic crystallites which are aligned along the longitudinal axis of the fibre; this is achieved through specialised spinning techniques (dry-jet wet spinning) and the use of liquid crystalline spinning dopes.8,10,23 Aggregated graphene, or graphite, has a distinctive WAXD diffraction peak at 26.6° 2θ – corresponding to the 002 Bragg reflection of stacked graphite sheets;24 this peak is not seen in fully exfoliated graphene.4,25,26 A 002 peak at 26° can be seen in the B7-PPTA-M25 fibres with a graphene loading of 2.5% or greater (Fig. 3a), however is not observed in any of the B7-PPTA-C750 fibres. This suggests that C750 graphene is relatively well-dispersed within the fibres and has not re-aggregated into GNPs during the spinning process – even at the relatively high loading of 10% graphene relative to PPTA.5
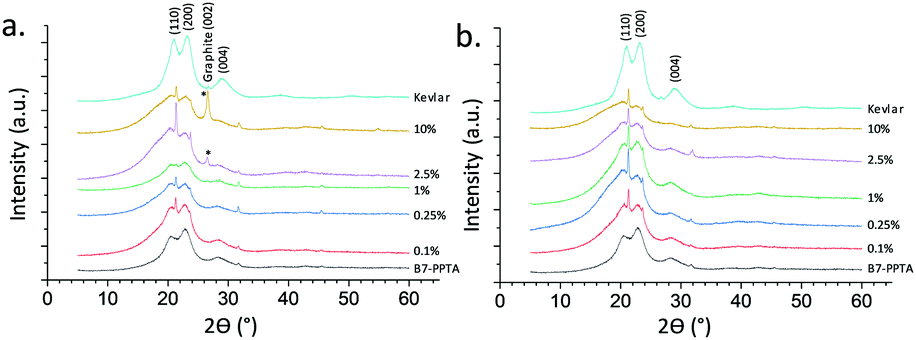 | ||
| Fig. 3 WAXD patterns for B7-PPTA with increasing loading of (a) M25 and (b) C750 graphene with Kevlar for comparison. Unassigned peaks are due to sample holder and adherent (petroleum jelly) background. | ||
Raman spectroscopy was employed to further analyse the graphene dispersion within the fibres. Raman spectroscopy is a common technique for the characterisation of graphene since it can differentiate between single and multi-layer sheets through characteristic changes to peaks around 2700 cm−1.27,28 These peaks were not observed however, which was attributed to the vibrational modes being diminished and attenuated through interactions with the PPTA matrix, meaning differentiation between single- and multi-layer graphene through Raman spectroscopy could not be performed in this instance (Fig. S4, ESI†).
The outstanding mechanical properties of commercial aramid fibres is largely due to a high degree of non-covalent intramolecular bonding, namely H-bonding between adjacent amide groups and π–π interactions.7 Restricted rotation around the amide bonds also promotes these interactions and the formation of semi-crystalline domains.10 It was hypothesised that the highly linear nature of PPTA polymers, coupled with their high degree of aromaticity, could effectively stabilise graphene sheets though significant π–π interactions. These interactions could also be effective in transferring stresses from the polymer matrix to the reinforcing graphene.11–14 Furthermore, alignment of the PPTA crystallites along the axis of the fibre (achieved in commercial aramid fibres through dry-jet wet spinning of liquid crystalline dopes) could also orientate the graphene sheets which may further improve mechanical properties and electrical conductivity.10,29 A basic computational model, based on the crystal structure of PPTA8 and a monolayer of graphene, was constructed which confirmed the possibility of π–π bond interactions between overlapping aromatic groups (Fig. S5, ESI†).
The mechanical properties of the fibres were assessed through uniaxial tensile testing (Fig. 4), however brittleness of the fibres under compression made loading samples into the testing instrument (i.e., securely clamping the fibres) and therefore obtaining reliable measurements difficult. The cross-sectional area of the fibres, required to calculate the ultimate tensile strength, was determined through a combination of cross-sectional SEM imaging and visible light microscopy (i.e., measurement of fibre diameter to calculate area). The cross-sectional SEM images of the fibres revealed large cavities (ca. 90% cavity volume) within the fibres, which likely compromised their mechanical integrity (Fig. S6, ESI†). These cavities were attributed to the release of gasses (e.g., H2O, HCl vapour) as CSA contacted the water coagulation bath – resulting in a degree of foaming at the point of fibre formation. N2 gas sorption and subsequent BET surface area analysis indicated a specific surface area of 16 m2 g−1 (Fig. S7, ESI†), suggesting a degree of non-visible micro-/meso porosity in addition to the larger cavities. It should be noted that voids within commercial Kevlar fibres (i.e., PPTA spun from anhydrous sulfuric acid) have also been observed,10,30 and have been attributed to differences in the coagulation rates between the skin and core of the fibres,31 as well as cavities caused by the presence of Na2SO4 (from the neutralisation of residual H2SO4).32 Other polymeric fibres such as poly(acrylonitrile) wet-spun from standard solvents (e.g. dimethyl sulfoxide, dimethylformamide) into water also have significant voids arising from phase separation, which can be removed by optimising the coagulation bath composition/temperature, as well as post-spinning fibre treatment including drawing and heating under tension.33 It's therefore feasible that a more advanced fibre spinning line could overcome the issue of cavities within the fibres and hence improve the mechanical properties of the fibres. The conductivity of the fibres was measured through a 4-probe technique. However, there was no detectable electrical conductivity within the sensitivity range of the instrument. The lack of conductivity was attributed to the pores and cavities within the fibres impeding electron percolation.
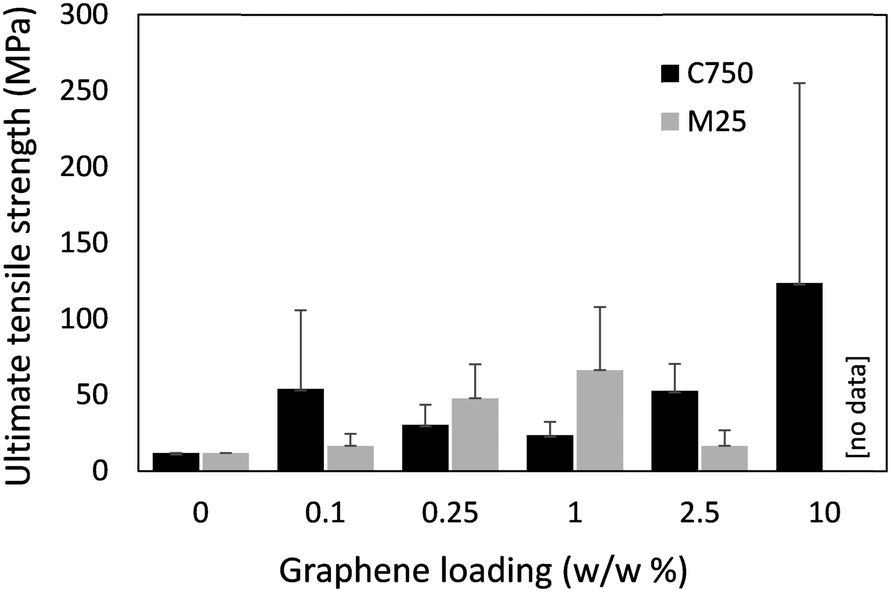 | ||
| Fig. 4 Ultimate tensile strength of B7-PPTA with increasing amounts of M25 and C750 graphene. Note, fibres with 10% loading of M25 were too brittle to measure and therefore no data could be obtained. | ||
The fact that CSA can effectively dissolve PPTA as well as produce stable colloidal dispersions of single-layer graphene1 was exploited to produce PPTA–graphene composite fibres. Graphene sheets derived from C750 GNPs (average diameter of 2 μm) were effectively dispersed throughout the PPTA fibres up to a loading of 10% by mass without any significant aggregation as determined by WAXD and cross-polarised light microscopy. Aggregation was, however, observed for graphene sheets derived from M25 (average diameter of 25 μm) at a loading of 2.5% and greater. Significant π–π interactions between PPTA chains and graphene sheets may facilitate homogenous dispersion throughout the polymer matrix and effectively distribute mechanical stresses. Gasses released at the point of fibre formation resulted in porous fibres with large cavities, compromising the mechanical and electrical conductivity properties. Further optimisation of PPTA synthesis to increase Mave, development of the spinning rig, and post-fibre treatment could overcome these issues to produce non-porous aramid fibres with a high loading of well-dispersed graphene. This superacid co-processing approach could also be applied to other carbon nanomaterials (e.g., single or multi-walled carbon nanotubes, fullerenes, activated carbon particles, etc.) or other acid-resistant polymers.
This work was funded by the Defence Science and Technology Laboratory and the Engineering and Physical Sciences Research Council (EPSRC; grant EP/N025504/1). The work is a contribution from the EPSRC/BBSRC Future Biomanufacturing Research Hub (EP/S01778X/1).
Conflicts of interest
There are no conflicts to declare.Notes and references
- N. Behabtu, J. R. Lomeda, M. J. Green, A. L. Higginbotham, A. Sinitskii, D. V. Kosynkin, D. Tsentalovich, A. N. G. Parra-Vasquez, J. Schmidt, E. Kesselman, Y. Cohen, Y. Talmon, J. M. Tour and M. Pasquali, Nat. Nanotechnol., 2010, 5, 406 CrossRef CAS.
- C. Lee, X. Wei, J. W. Kysar and J. Hone, Science, 2008, 321, 385–388 CrossRef CAS.
- D. W. Johnson, B. P. Dobson and K. S. Coleman, Curr. Opin. Colloid Interface Sci., 2015, 20, 367–382 CrossRef CAS.
- H. Kim, A. A. Abdala and C. W. Macosko, Macromolecules, 2010, 43, 6515–6530 CrossRef CAS.
- J. R. Potts, D. R. Dreyer, C. W. Bielawski and R. S. Ruoff, Polymer, 2011, 52, 5–25 CrossRef CAS.
- S. Stankovich, D. A. Dikin, G. H. B. Dommett, K. M. Kohlhaas, E. J. Zimney, E. A. Stach, R. D. Piner, S. T. Nguyen and R. S. Ruoff, Nature, 2006, 442, 282–286 CrossRef CAS PubMed.
- E. G. Chatzi and J. L. Koenig, Polym.-Plast. Technol. Eng., 1987, 26, 229–270 CrossRef CAS.
- M. G. Northolt and J. J. Van Aartsen, J. Polym. Sci., Polym. Lett. Ed., 1973, 11, 333–337 CrossRef CAS.
- H. Blades, US Pat., US3869429A, 1975 Search PubMed.
- Z. Hongpeng, K. Haijuan, L. Jing, M. Yua, D. Ahmed, Z. Hongpeng, K. Haijuan, L. Jing, M. Yu and Y. Muhuo, Mater. Res., 2014, 17, 1180–1200 CrossRef.
- Y. Xu, H. Bai, G. Lu, C. Li and G. Shi, J. Am. Chem. Soc., 2008, 130, 5856–5857 CrossRef CAS PubMed.
- Q. Su, S. Pang, V. Alijani, C. Li, X. Feng and K. Müllen, Adv. Mater., 2009, 21, 3191–3195 CrossRef CAS.
- K. W. J. Heard, C. Bartlam, C. D. Williams, J. Zhang, A. A. Alwattar, M. S. Little, A. V. S. Parry, F. M. Porter, M. A. Vincent, I. H. Hillier, F. R. Siperstein, A. Vijayaraghavan, S. G. Yeates and P. Quayle, ACS Omega, 2019, 4, 1969–1981 CrossRef CAS.
- J. Liu, L. Tao, W. Yang, D. Li, C. Boyer, R. Wuhrer, F. Braet and T. P. Davis, Langmuir, 2010, 26, 10068–10075 CrossRef CAS.
- W. Lu, S. Liu, X. Qin, L. Wang, J. Tian, Y. Luo, A. M. Asiri, A. O. Al-Youbi and X. Sun, J. Mater. Chem., 2012, 22, 8775–8777 RSC.
- İ. Mutlay and L. B. Tudoran, Fullerenes, Nanotubes, Carbon Nanostruct., 2013, 21, 149–157 CrossRef.
- R. J. W. Cremlyn, Chlorosulfonic Acid: A Versitile Reagent, Royal Society of Chemistry, 1st edn, 2002 Search PubMed.
- B. H. Bersted and T. G. Anderson, J. Appl. Polym. Sci., 1990, 39, 499–514 CrossRef CAS.
- H. Blades, US Pat., US3767756A, 1973 Search PubMed.
- H. Bin, Li, W. Y. Shi and Y. F. Zhang, Adv. Mater. Res., 2014, 941, 1249–1252 Search PubMed.
- P. Wang, K. Wang, J. Zhang and G. Luo, RSC Adv., 2015, 5, 64055–64064 RSC.
- J. G. Um, Y.-S. Jun, H. Alhumade, H. Krithivasan, G. Lui and A. Yu, RSC Adv., 2018, 8, 17091–17100 RSC.
- T.-M. Wu and J. Blackwell, Macromolecules, 1996, 29, 5621–5627 CrossRef CAS.
- A. Milev, M. Wilson, G. S. K. Kannangara and N. Tran, Mater. Chem. Phys., 2008, 111, 346–350 CrossRef CAS.
- M. J. McAllister, J.-L. Li, D. H. Adamson, H. C. Schniepp, A. A. Abdala, J. Liu, M. Herrera-Alonso, D. L. Milius, R. Car, R. K. Prud’homme and I. A. Aksay, Chem. Mater., 2007, 19, 4396–4404 CrossRef CAS.
- H. C. Schniepp, J.-L. Li, M. J. McAllister, H. Sai, M. Herrera-Alonso, D. H. Adamson, R. K. Prud’homme, R. Car, D. A. Saville and I. A. Aksay, J. Phys. Chem. B, 2006, 110, 8535–8539 CrossRef CAS.
- A. C. Ferrari and D. M. Basko, Nat. Nanotechnol., 2013, 8, 235 CrossRef CAS.
- A. C. Ferrari, Solid State Commun., 2007, 143, 47–57 CrossRef CAS.
- T. D. Fornes and D. R. Paul, Polymer, 2003, 44, 4993–5013 CrossRef CAS.
- M. G. Dobb, D. J. Johnson, A. Majeed and B. P. Saville, Polymer, 1979, 20, 1284–1288 CrossRef CAS.
- M. Panar, P. Avakian, R. C. Blume, K. H. Gardner, T. D. Gierke and H. H. Yang, J. Polym. Sci., Polym. Phys. Ed., 1983, 21, 1955–1969 CrossRef CAS.
- M. Grujicic, P. S. Glomski, B. Pandurangan, W. C. Bell, C.-F. Yen and B. A. Cheeseman, J. Mater. Sci., 2011, 46, 4787–4802 CrossRef CAS.
- E. Morris, M. Weisenberger and G. Rice, Fibers, 2015, 3, 560–574 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and supplementary figures. See DOI: 10.1039/c9cc04548a |
| This journal is © The Royal Society of Chemistry 2019 |