
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIcosahedral carboranes as scaffolds for congested regioselective polyaryl compounds: the distinct distance tuning of C–C and its antipodal B–B†
Zsolt
Kelemen
 a,
Ariadna
Pepiol
a,
Marius
Lupu
a,
Reijo
Sillanpää
b,
Mikko M.
Hänninen
b,
Francesc
Teixidor
a and
Clara
Viñas
*a
a,
Ariadna
Pepiol
a,
Marius
Lupu
a,
Reijo
Sillanpää
b,
Mikko M.
Hänninen
b,
Francesc
Teixidor
a and
Clara
Viñas
*a
aInstitut de Ciència de Materials de Barcelona, ICMAB-CSIC Campus U.A.B., 08193 Bellaterra, Spain. E-mail: clara@icmab.es
bDepartment of Chemistry, University of Jyväskylä, FIN-40351, Jyväskylä, Finland
First published on 25th June 2019
Abstract
Four-fold aryl substituted o-carborane derivatives with defined patterns of substitution at the antipodal region of the cluster carbon atoms have been achieved. It is proven that this region is congested but lacks steric hindrance. Also, the two antipodal sites Cc–Cc and B9–B12 are affected very distinctly by electron donor substituents.
The closo C2B10H12 icosahedral carboranes, o-(1,2), m-(1,7), or p-(1,12), are the most widely studied boron clusters; they can be functionalized by different reactions,1 in a regioselective manner, with many possible sites of substitution. o-Carborane, 1,2-closo-C2B10H12, acts as a strong electron-withdrawing molecule through the substitution on the carbon, Cc,2 and as an electron-donating moiety through the boron vertices with a gradation depending on the distance to the carbon atoms.3 On the other hand, it is well known that the Cc–Cc bond length in o-carborane and metalla-o-carborane strongly depends on the electronic nature of the immediate Cc-substituents;4 π-donor substituents bonded to the Cc, as well as atoms with lone pairs, donate electron density to the σ*(Cc–Cc) antibonding orbital (negative hyperconjugation) of o-carborane, which lies in the Cc–Cc bond, increasing significantly the Cc–Cc bond distance.5 In the case of aryl substituents, the orientation of the substituents has a significant impact on the electron donation.6 The longest Cc–Cc distance ever reported is 2.156(4) Å for the 1,2-(CR2Fc)2-1,2-closo-C2B10H10, {R = pentamethylene; Fc = (η5-C5H4)Fe(η5-C5H5)} o-carborane cluster derivative.7 While the plasticity of the Cc–Cc bond has been investigated, the possibility of a similar behaviour at the B–B bonds located antipodal to the cluster carbon atoms remained unexplored.
In addition, if π-aromaticity and tridimensional aromaticity of the icosahedral boranes are two sides of the same coin,8 could it be possible to obtain hybrid polyaryl compounds by merging the 2D organic aromatic groups at the dense antipodal region of the cluster carbon atoms with 3D inorganic icosahedral clusters?
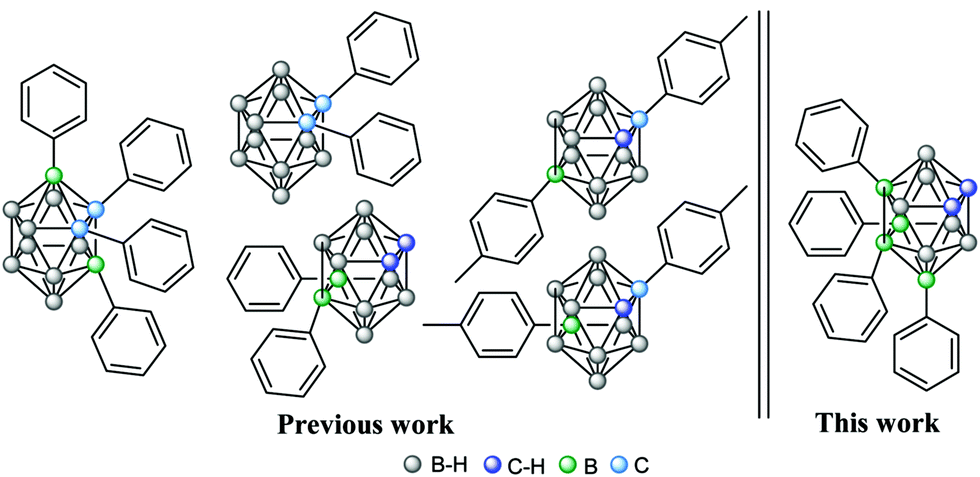
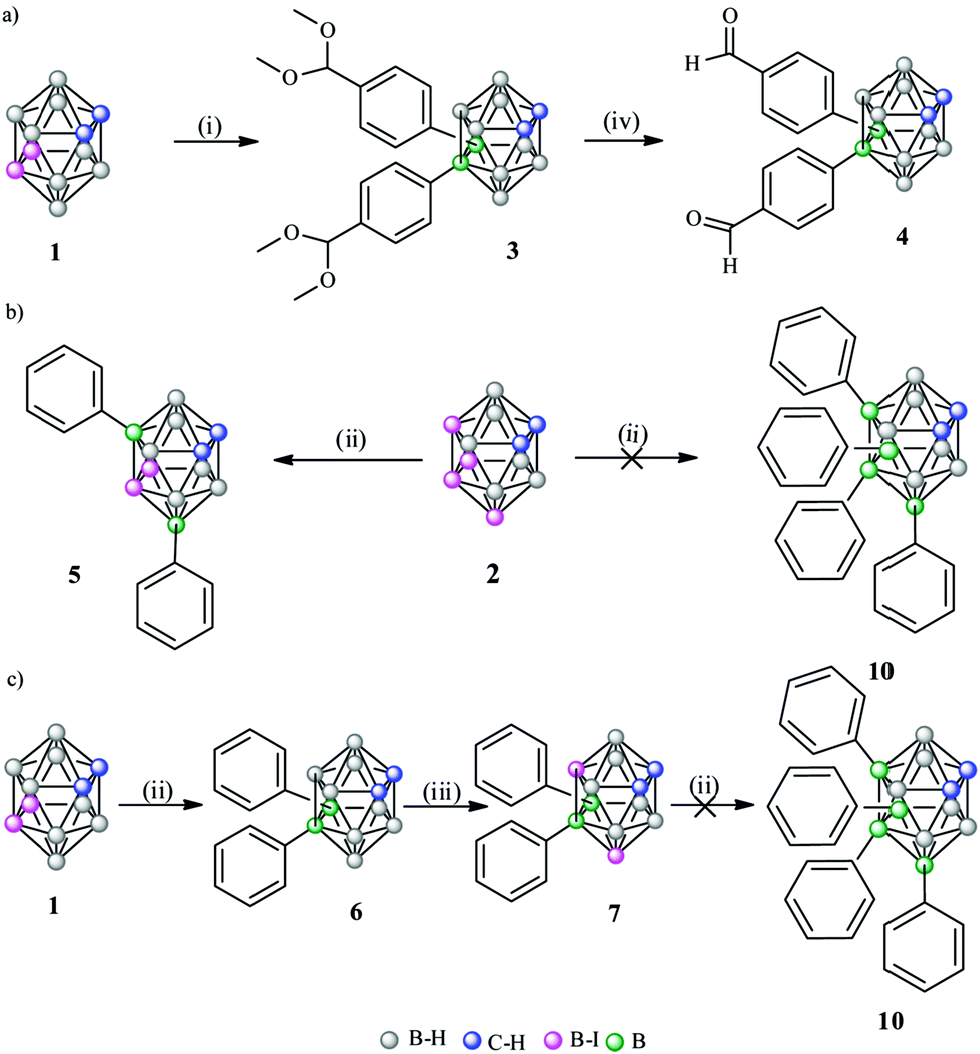
To produce B–C substitutions, a useful and general method is by electrophilic iodination of o-carborane followed by Kumada cross-coupling reaction.9 This implies that one should start from the appropriate iodocarborane derivatives. A less regioselective B(8)/B(9)-aryl-o-carborane Pd-catalysed monoarylation with aryl iodides via B–H activation was reported,10 as well as a palladium-catalysed regioselective diarylation on B(4,5) of o-carborane directly from B–H with aryl halides, with the help of the traceless directing carboxylic group.11 With hindered substitutions on the neighbouring C/B atoms, 3-Ph-1,2-Ph2-1,2-closo-C2B10H9 and 3,6-Ph2-1,2-Ph2-1,2-closo-C2B10H912 derivatives were obtained in low yield by using the sequential nucleophilic-capping reactions of BPhCl2 in the precursors [7,8-Ph2-7,8-nido-C2B9H9]2− and [6-Ph-7,8-Ph2-7,8-nido-C2B9H8]2−. It should be noted that disubstituted 9,12-Ph2-1,2-closo-C2B10H10,13 1,9- and 1,12-(p-MeC6H4)2-1,2-closo-C2B10H10 were also reported.14 We interpret that the presence of the four phenyl groups in the crowded area adjacent to the Cc vertices is accessible because of the different behaviour of all the atoms connected to the C2B2 region where the strong electron-withdrawing (Cc) and electron-donating moieties (B) of the o-carboranyl group co-exist. As mentioned, it was our main target to elucidate if the four iodo groups in the compact and electron-rich region of the o-carborane cluster (Chart 1) could be fully or only partially replaced by the organic aryl (sp2) groups such as phenyl to produce a “four phenyl congested site”. Furthermore, it could provide a good picture of the B–B tuning possibility to the antipodal Cc–Cc unit. This would be a potentially useful heavily congested electron-rich region due to the regioselective formation of B–C(sp2), B–C(sp) and B–C(sp3). To achieve so, the cross-coupling reaction on the B-iodinated o-carboranes with Grignard reagents in the presence of the Pd(II) and Cu(I) catalysts was studied. Two were the starting iodinated derivatives, 9,12-I2-1,2-closo-C2B10H10 (1) and 8,10,9,12-I4-1,2-closo-C2B10H8 (2). In 2, the four iodo groups occupy a highly dense region antipodal to the Cc–Cc atoms. In 1, only two adjacent iodo groups are present, again antipodal to the Cc–Cc unit. Four Grignard reagents were used, phenylethynyl magnesium, phenyl magnesium and allyl magnesium chlorides, and 4-benzaldehyde dimethyl acetal magnesium bromide. The first, the second and the fourth would produce a highly electron dense and congested region in o-carborane derivatives, whereas the third would produce a more relaxed space. In a typical experiment, 1 and cis-[PdCl2(PPh3)2] and CuI as catalysts were dispersed in anhydrous THF and treated with the appropriate Grignard reagent at low temperatures (Scheme 1). After refluxing overnight, the corresponding crude compounds were purified (see ESI†). The cross-coupling reaction of 1 with 4-benzaldehyde dimethyl acetal magnesium bromide proceeded to give 9,12-(C6H4CH(OCH3)2)2-1,2-closo-C2B10H10, 3, in 71% yield (Scheme 1a), which was confirmed by multinuclear NMR, FTIR, mass spectrometry and elemental analysis.
 | ||
| Chart 1 Different polyaryl-o-carboranes. | ||
 | ||
| Scheme 1 Kumada cross coupling reactions on 1 (a) and 2 (b) using cis-[Pd(PPh3)2Cl2] and CuI as catalysts, in THF refluxing overnight. (i) 4-Benzaldehyde dimethyl acetal magnesium bromide, (ii) PhMgCl,13 (iii) solvent free electrophilic iodination with I2 (10 eq.) at 210 °C during 4 h. (iv) Extraction with Et2O in 3 M aqueous solution HCl. | ||
Deprotection of the carbonyl group was achieved with diethylether in aqueous HCl 3 M solution to produce 9,12-(C6H4CHO)2-1,2-closo-C2B10H10, 4, in 96% yield (Scheme 1a). Suitable crystals for X-ray diffraction of 3 and 4, which were grown in chloroform (Fig. 1) unambiguously and unequivocally confirmed that 3 and 4 were obtained. Furthermore, tetrasubstitution on 2 to yield 8,9,10,12-Ph4-1,2-closo-C2B10H8 (10) was expected by reaction of 2 with phenyl magnesium chloride but 8,10-Ph2-9,12-I2-1,2-closo-C2B10H8 (5) was obtained with only two substitutions (Scheme 1b).9d To get more insight into the reason for this atypical or at least unexpected result, we investigated 11B{1H}-NMR chemical shifts (Fig. 2) and performed DFT calculations. The 11B{1H}-NMR chemical shifts of 2 were unambiguously assigned9d with the aid of a 2D 11B{1H}–11B{1H}-NMR COSY experiment.15 The B(9,12) resonances of these B–I vertices at 2 appear at −7.1 ppm that are not so different from the B–H resonances of the unreactive B(9,12) vertices in the pristine 1,2-closo-C2B10H12 (−3.1 ppm); on the contrary, the B(8,10) vertex resonances of 2 appear close to −16 ppm being very similar to the B–I reactive B(9,12) vertices in 1 that resonate at −14.9 ppm (Fig. 2). It can be seen in the 11B{1H}-NMR of 1,2-closo-C2B10H12, 9,12-I2-closo-1,2-C2B10H10 (1) and 8,10,9,12-I4-1,2-closo-C2B10H8 (2) that the four iodo groups in the congested area of 2 are not electronically identical. Electronically, the B(8,10) vertices in 2 are strongly affected upon I substitution shifting 12 ppm upfield from B(8,10)–H to B(8,10)–I. These B(8,10)–I vertices are highly reactive and lead readily to disubstitution to obtain B–Ph in 5. The calculated Mulliken charges (Table S1 in ESI†) are in good agreement with the chemical shifts; the charge of the reactive B(8,10)–I vertices in 2 is +0.66, while the charge of the B(8,10)–I vertices is +0.31 in 5.
 | ||
| Fig. 1 Crystal structures of 3 (left) and 4 (right). Thermal ellipsoids have been drawn with 20% probability. The B9–B12 distances are 1.804(4) and 1.798(3) Å for 3 and 4. | ||
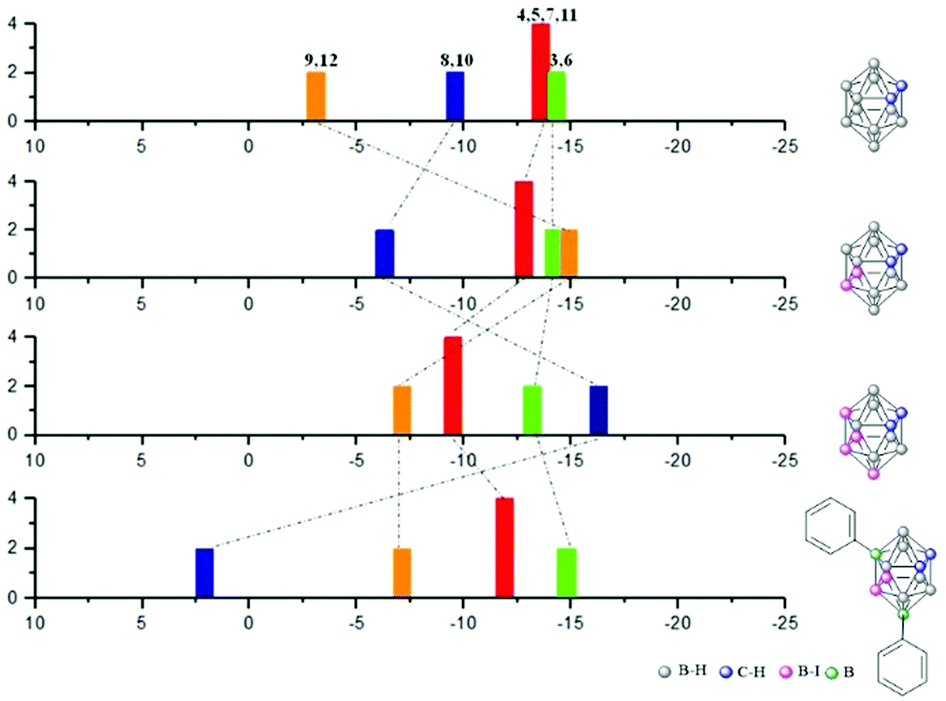 | ||
| Fig. 2 Diagram 11B{1H}-NMR spectra with the peak assignments for the o-carborane, 1, 2 and 5 derivatives. | ||
Clearly, the B(9,12) vertices in 5 are not only less reactive electronically, but less accessible for the reactant (steric hindrance) due to the phenyl substituents at the B(8,10) vertices. Since the four-fold phenylation of 2 was unsuccessful, a new synthetic strategy aiming at 8,9,10,12-Ph4-1,2-closo-C2B10H8 was designed. The positional isomer of 5, 8,10-I2-9,12-Ph2-1,2-closo-C2B10H8 (7) was synthesised starting from 6. In 7 the two iodo substituents would be ready for substitution at less crowded positions than in 5; moreover, DFT calculations suggest a more reactive B–I vertex. This led us to perform the cross coupling reaction of 2 with PhMgCl to obtain 9,12-Ph2-1,2-closo-C2B10H10, 6.13 This was treated with I2 in a sealed tube at 210 °C for 4 h (73% yield, Scheme 1c).
The 11B NMR spectra of 7 verified the more reactive B–I vertices (they resonate at −20.1 ppm), in agreement with the DFT calculations. Furthermore, as it was mentioned earlier, the different allocation of the four substituents in 7 with respect to 5 could decrease the steric effect of the two bulky phenyl groups since they are sited at the neighbouring vertices B(9,12), and the two remaining iodo substituents are placed one far from the other and at the lateral position, so the B–I substitutions by the phenyl groups are sterically feasible. However, the cross-coupling reaction of 7 with PhMgCl did not lead to the sought four-fold phenylation, only to unreacted 7 (Scheme 1c).
To check the possibility of further substitution on the 5 congested electron-rich region of the o-carborane cluster, we decided to combine the pre-existing sp2 (Ph) in 5, with the sp (PhC![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C) and sp3 (allyl) units, respectively. The reaction of 5 with phenylethynyl magnesium chloride to produce 8 was unsuccessful as no iodo was exchanged (Scheme 2). Instead, when using a less crowded allyl magnesium chloride, the coupling reaction of 5 with allyl magnesium chloride leads to the four-fold substitution by the formation of 8,10-Ph2-9,12-(CH2CHCH2)2-1,2-closo-C2B10H8, 9, in 75% yield (Scheme 2). 11B and 1H NMR spectroscopy, MALDI-TOF-MS and EA data along with its X-ray structure from the crystals grown from a chloroform solution (Fig. 3 and ESI†) confirmed the allylation of B(9,12) vertices on 5. Since the sp and sp2 four-fold substitution is possible, the steric hindrance in the proposed tetra-substituted 8,10,9,12-Ph4-1,2-closo-C2B10H8 (10, Scheme 3) compound was investigated by using different hypothetical isodesmic reactions (see more details in ESI†). The DFT calculations indicated that there is not enough steric hindrance in 10 to prevent four-fold phenyl substitution, therefore 10 should be thermodynamically available and so its synthesis should be restricted only by kinetic reasons. To prove this hypothesis, we have modified the reaction conditions by using 1,4-dioxane instead of THF, which increases the reaction temperature up to 101 °C (Scheme 3). After 16 hours of refluxing, the 1H-NMR spectra of the crude mixture displayed new broad peaks in the region 4.5–5.5 ppm, in which the Cc–H resonates, indicating the formation of additional compounds.3a By working up (see ESI†), the isolation of four-fold substituted o-carborane derivative 10 as well as the trisubstituted 11 was achieved in 18 and 59% yield, respectively. To further increase the yield of 10, mesitylene (b.p. 165 °C) and diglyme (b.p. 162 °C) were used as solvents however, the yield was lower (12% and 16%, respectively) probably due to other side reactions. Finally, the reaction in toluene, slightly increased the yield of 10 to 21 and 34% after 16 h and 5 days’ reaction time, respectively. The reaction in toluene was carried out under microwave irradiation (see in ESI†) as well, but a similar ratio of 10 and 11 (comparing the reaction in toluene after 16 h reflux) was observed after 2 h irradiation at 120 °C according to the 1H NMR spectra of the crude product. It is to be emphasized that the by-product of 10 (compound 11) is suitable for recycling, thus the overall yield can be significantly improved. Crystals of 10 and 11 suitable for X-ray diffraction (Fig. 4) were obtained using the vapour diffusion technique (pentane/acetone, see ESI†). The four-fold phenyl groups at the dense antipodal region of the cluster carbon atoms of the closo-o-carborane in 10 parallels the nanohybrid [η5-C60Ph5]−,16a that made the extremely hydrophobic C60 fullerene soluble in water.16b
C) and sp3 (allyl) units, respectively. The reaction of 5 with phenylethynyl magnesium chloride to produce 8 was unsuccessful as no iodo was exchanged (Scheme 2). Instead, when using a less crowded allyl magnesium chloride, the coupling reaction of 5 with allyl magnesium chloride leads to the four-fold substitution by the formation of 8,10-Ph2-9,12-(CH2CHCH2)2-1,2-closo-C2B10H8, 9, in 75% yield (Scheme 2). 11B and 1H NMR spectroscopy, MALDI-TOF-MS and EA data along with its X-ray structure from the crystals grown from a chloroform solution (Fig. 3 and ESI†) confirmed the allylation of B(9,12) vertices on 5. Since the sp and sp2 four-fold substitution is possible, the steric hindrance in the proposed tetra-substituted 8,10,9,12-Ph4-1,2-closo-C2B10H8 (10, Scheme 3) compound was investigated by using different hypothetical isodesmic reactions (see more details in ESI†). The DFT calculations indicated that there is not enough steric hindrance in 10 to prevent four-fold phenyl substitution, therefore 10 should be thermodynamically available and so its synthesis should be restricted only by kinetic reasons. To prove this hypothesis, we have modified the reaction conditions by using 1,4-dioxane instead of THF, which increases the reaction temperature up to 101 °C (Scheme 3). After 16 hours of refluxing, the 1H-NMR spectra of the crude mixture displayed new broad peaks in the region 4.5–5.5 ppm, in which the Cc–H resonates, indicating the formation of additional compounds.3a By working up (see ESI†), the isolation of four-fold substituted o-carborane derivative 10 as well as the trisubstituted 11 was achieved in 18 and 59% yield, respectively. To further increase the yield of 10, mesitylene (b.p. 165 °C) and diglyme (b.p. 162 °C) were used as solvents however, the yield was lower (12% and 16%, respectively) probably due to other side reactions. Finally, the reaction in toluene, slightly increased the yield of 10 to 21 and 34% after 16 h and 5 days’ reaction time, respectively. The reaction in toluene was carried out under microwave irradiation (see in ESI†) as well, but a similar ratio of 10 and 11 (comparing the reaction in toluene after 16 h reflux) was observed after 2 h irradiation at 120 °C according to the 1H NMR spectra of the crude product. It is to be emphasized that the by-product of 10 (compound 11) is suitable for recycling, thus the overall yield can be significantly improved. Crystals of 10 and 11 suitable for X-ray diffraction (Fig. 4) were obtained using the vapour diffusion technique (pentane/acetone, see ESI†). The four-fold phenyl groups at the dense antipodal region of the cluster carbon atoms of the closo-o-carborane in 10 parallels the nanohybrid [η5-C60Ph5]−,16a that made the extremely hydrophobic C60 fullerene soluble in water.16b
 | ||
Scheme 2 Kumada cross coupling reactions on 2 by using (i) PhCCMgCl, (ii) CH2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHCH2MgCl, in the presence of cis-[PdCl2(PPh3)2] and CuI as catalysts, in refluxing THF overnight. CHCH2MgCl, in the presence of cis-[PdCl2(PPh3)2] and CuI as catalysts, in refluxing THF overnight. | ||
 | ||
| Fig. 3 Crystal structures of 9. Thermal ellipsoids have been drawn with 20% probability. | ||
 | ||
| Scheme 3 Kumada cross coupling reactions on 2 in refluxing dioxane for 16 h. | ||
 | ||
| Fig. 4 Crystal structures of 10 (right) and 11 (left). Thermal ellipsoids have been drawn with 20% probability. | ||
A search at the Cambridge Structural Database17 shows only seven hits for 9,12-(aryl)2-1,2-closo-C2B10B10, whose B9–B12 distances are in the range 1.781–1.824 Å (Table S1 in ESI†), which are comparable, although a bit longer than that in o-carborane (1.776 Å).18 Although a certain plasticity at the B–B bonds antipodal to the Cc atoms is observed, the effect is considerably smaller than the plasticity found for the adjacent Cc atoms in the o-carborane cluster. This is not surprising as the antibonding orbital is mainly located between the adjacent Cc atoms for these derivatives.5 Compared to the electron-donation to the σ*(Cc–Cc), whose extent in terms of distance has been found experimentally at 2.156(4) Å in 1,2-(CR2Fc)2-1,2-closo-C2B10H107 and computed at 2.64 Å for 1,2-(CH2−)2-1,2-C2B10H10,5 the maximum elongation found in the antipodal region of 10 (1.822 A) or 11 (1.824) indicates a minor effect, near 0.048 Å. In agreement, the second-order perturbation theory analysis on the NBO basis reveals weak interactions (sum of them ∼12 kcal mol−1) between the aryl π-systems and the antibonding orbitals of the boron atoms, which was in good agreement with enlargement of the B–B distances in the cluster. This enforces the concept of the electron back donation to the σ*(Cc–Cc) vs. the very much abused steric hindrance. The proven, although weak, influence of the aryl groups in the plasticity on the B9–B12 bonds and the possibility of substitution at the Cc–H vertices foresee these o-carborane derivatives as appropriate synthons for surface functionalization; research in this direction is underway in our laboratories.
This work has been supported by the Spanish Ministerio de Economía y Competitividad (CTQ2016-75150-R), the Generalitat de Catalunya (2017SGR1720) and European Union's Horizon 2020 Marie Skłodowska-Curie grant agreement MSCA-IF-2016-751587.
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) R. N. Grimes, Carboranes, Elsevier Inc, New York, 3rd edn, 2016 Search PubMed; (b) P. C. Andrews, R. J. Hardie and C. L. Raston, Coord. Chem. Rev, 1999, 189, 169–198 CrossRef CAS; (c) J. F. Valliant, K. J. Guenther, A. S. King, P. Morel, P. Schaffer, O. O. Sogbein and K. A. Stephenson, Coord. Chem. Rev., 2002, 232, 173–230 CrossRef CAS; (d) T. J. Wedg and M. F. Hawthorne, Coord. Chem. Rev., 2003, 240, 111–128 CrossRef; (e) M. A. Fox, Polyhedral Carboranes, in Comprehensive Organometallic Chemistry III, ed. R. H. Crabtree and D. M. P. Mingos, Elsevier, Oxford, 2007, ch. 3.02, vol. 3, pp. 49–112 Search PubMed; (f) V. N. Kalinin and V. A. Ol'shevskaya, Russ. Chem. Bull. Int. Ed., 2008, 57, 815–836 CrossRef CAS; (g) I. B. Sivaev and V. V. Bregadze, Eur. J. Inorg. Chem., 2009, 1433–1450 CrossRef CAS; (h) F. Issa, M. Kassiou and L. M. Rendina, Chem. Rev., 2011, 111, 5701–5722 CrossRef CAS PubMed; (i) M. Scholz and E. Hey-Hawkins, Chem. Rev., 2011, 111, 7035–7062 CrossRef CAS PubMed; (j) In Boron Science, ed. N. S. Hosmane, CRC Press, New York, 2012 Search PubMed; (k) D. Olid, R. Nuñez, C. Viñas and F. Teixidor, Chem. Soc. Rev., 2013, 42, 3318–3336 RSC.
- (a) F. Teixidor, R. Núñez, C. Viñas, R. Sillanpää and R. Kivekäs, Angew. Chem., Int. Ed., 2000, 39, 4290–4292 CrossRef CAS; (b) R. Núñez, P. Farràs, F. Teixidor, C. Viñas, R. Sillanpää and R. Kivekäs, Angew. Chem., Int. Ed., 2006, 45, 1270–1272 CrossRef PubMed.
- (a) F. Teixidor, G. Barbera, A. Vaca, R. Kivekäs, R. Sillanpää, J. Oliva and C. Viñas, J. Am. Chem. Soc., 2005, 127, 10158–10159 CrossRef CAS PubMed; (b) A. M. Spokoyny, C. W. Machan, D. J. Clingerman, M. S. Rosen, M. J. Wiester, R. D. Kennedy, C. L. Stern, A. A. Sarjeant and C. A. Mirkin, Nat. Chem., 2011, 3(8), 590–596 CrossRef CAS PubMed.
- F. Teixidor and C. Viñas, Science of Synthesis, Houben-Weyl Methods of Molecular Transformations, ed. D. E. Kaufmann and D. S. Matteson, Georg Thieme Verlag, Stuttgart-New York, 2005, vol. 6, pp. 1235–1267 Search PubMed.
- J. M. Oliva, N. L. Allan, P. v. R. Schleyer, C. Viñas and F. Teixidor, J. Am. Chem. Soc., 2005, 127, 13538–13547 CrossRef CAS PubMed.
- (a) E. S. Alekseyeva, M. A. Fox, J. A. K. Howard, J. A. H. MacBride and K. Wade, Appl. Organomet. Chem., 2003, 17, 499–508 CrossRef CAS; (b) I. V. Glukhov, M. Y. Antipin and K. A. Lyssenko, Eur. J. Inorg. Chem., 2004, 1379–1384 CrossRef CAS.
- B. W. Hutton, F. MacIntosh, D. Ellis, F. Herisse, S. A. Macgregor, D. McKay, V. Petrie-Armstrong, G. M. Rosair, D. S. Perekalin, H. Tricas and A. J. Welch, Chem. Commun., 2008, 5345–5347 RSC.
- J. Poater, M. Solà, C. Viñas and F. Teixidor, Angew. Chem., Int. Ed., 2014, 53, 12191–12195 CrossRef CAS PubMed.
- (a) G. Harakas, T. Vu, C. B. Knobler and M. F. Hawthorne, J. Am. Chem. Soc., 1998, 120, 6405–6406 CrossRef CAS; (b) H. Lee, C. B. Knobler and M. F. Hawthorne, Chem. Commun., 2000, 2485–2486 RSC; (c) A. Himmelspach and M. Finze, Eur. J. Inorg. Chem., 2010, 2012–2014 CrossRef CAS; (d) A. Vaca, F. Teixidor, R. Sillanpää, R. Kivekäs and C. Viñas, Chem. Commun., 2011, 47, 2252–2254 RSC; (e) F. Teixidor, R. Sillanpää, A. Pepiol, M. Lupu and C. Viñas, Chem. – Eur. J., 2015, 21, 12778–12786 CrossRef CAS PubMed.
- K. Cao, Y. Huang, J. Yang and J. Wu, Chem. Commun., 2015, 51, 7257–7260 RSC.
- Y. Quan and Z. Xie, Angew. Chem., Int. Ed., 2016, 55, 1295–1298 CrossRef CAS PubMed.
- G. F. Jin, J.-H. Hwang, J.-D. Lee, K.-R. Wee, I.-H. Suh and S. O. Kang, Chem. Commun., 2013, 49, 9398–9400 RSC.
- M. J. Bayer, A. Herzog, M. Diaz, G. A. Harakas, H. Lee, C. B. Knobler and M. F. Hawthorne, Chem. – Eur. J., 2003, 9, 2732–2744 CrossRef CAS PubMed.
- M. A. Fox and K. Wade, J. Mater. Chem., 2002, 12, 1301–1306 RSC.
- (a) T. L. Venable, W. C. Hutton and R. N. Grimes, J. Am. Chem. Soc., 1984, 106, 29–37 CrossRef CAS; (b) G. B. Jacobsen, D. G. Meina, J. H. Morris, C. Thomson, S. J. Andrews, D. Reed, A. J. Welch and D. F. Gaines, J. Chem. Soc., Dalton Trans., 1985, 1645–1654 RSC.
- (a) M. Sawamura, H. Iikura and E. Nakamura, J. Am. Chem. Soc., 1996, 118, 12850–12851 CrossRef CAS; (b) S. Zhou, Ch. Burger, B. Chu, M. Sawamura, N. Nagahama, M. Toganoh, U. E. Hackler, H. Isobe and E. Nakamura, Science, 2001, 291, 1944–1947 CrossRef CAS PubMed.
- On May 2nd 2019 J. Bruno, J. C. Cole, P. R. Edgington, M. Kessler, C. F. Macrae, P. McCabe, J. Pearson and R. Taylor, New software for searching the Cambridge Structural Database and visualizing crystal structures, Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 389–397 CrossRef PubMed.
- M. G. Davidson, T. G. Hibbert, J. A. K. Howard, A. Mackinnon and K. Wade, Chem. Commun., 1996, 2285–2286 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1562911, 1871937, 1871938, 1913782 and 1913783. Synthesis and characterization of 1–11. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9cc04526k |
| This journal is © The Royal Society of Chemistry 2019 |