
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Indirect state-of-charge determination of all-solid-state battery cells by X-ray diffraction†
Timo
Bartsch
*a,
A-Young
Kim
 a,
Florian
Strauss
a,
Lea
de Biasi
ab,
Jun Hao
Teo
a,
Jürgen
Janek
ac,
Pascal
Hartmann
ad and
Torsten
Brezesinski
*a
a,
Florian
Strauss
a,
Lea
de Biasi
ab,
Jun Hao
Teo
a,
Jürgen
Janek
ac,
Pascal
Hartmann
ad and
Torsten
Brezesinski
*a
aBattery and Electrochemistry Laboratory (BELLA), Institute of Nanotechnology (INT), Karlsruhe Institute of Technology (KIT), Hermann-von-Helmholtz-Platz 1, 76344 Eggenstein-Leopoldshafen, Germany. E-mail: timo.bartsch@kit.edu; torsten.brezesinski@kit.edu
bInstitute for Applied Materials-Energy Storage Systems (IAM-ESS), Karlsruhe Institute of Technology (KIT), Hermann-von-Helmholtz-Platz 1, 76344 Eggenstein-Leopoldshafen, Germany
cInstitute of Physical Chemistry & Center for Materials Science (ZfM/LaMa), Justus-Liebig-University Giessen, Heinrich-Buff-Ring 17, 35392 Giessen, Germany
dBASF SE, Carl-Bosch-Strasse 38, 67056 Ludwigshafen, Germany
First published on 26th August 2019
Abstract
Determining the state-of-charge of all-solid-state batteries via both ex situ and operando X-ray diffraction, rather than by electrochemical testing (may be strongly affected by electrically isolated/inactive material, irreversible side reactions, etc.), is reported. Specifically, we focus on bulk-type cells and use X-ray diffraction data obtained on a liquid electrolyte-based Li-ion cell as the reference standard for changes in lattice parameters with (de)lithiation.
In the electrochemical energy storage area, all-solid-state batteries (SSBs) are gaining much interest recently. Because SSBs seem to have the potential to deliver higher energy and power density than conventional Li-ion batteries (LIBs) using a liquid (organic) electrolyte, they may better meet the needs of the electric car industry. In addition, they promise improved safety and offer a wider operating temperature range than LIBs.1–3 Nonetheless, competitiveness in terms of performance, costs and so forth is still far from being reached. Moreover, several problems such as inherent instabilities at the cathode active material (CAM)/solid electrolyte (SE) interface, for example, and chemo-mechanical degradation as well as other challenges, especially with developing scalable fabrication processes, have yet to be overcome.3–6
One promising approach is the use of lithium thiophosphate SEs and high-energy CAMs (Ni-rich layered oxides),7–9 for which tailored interfacial design seems crucial in achieving stable cell performance. As a general rule, ionic and electronic percolation must be ensured to mitigate overvoltage and connectivity issues.8 However, decomposition of thiophosphate-based SEs during cycling and/or storage affects the stability of SSB cells strongly negatively.7 Applying a protective surface coating to the CAM particles is widely accepted as a means to address the latter problem by improving the interfacial stability (preventing direct contact between the CAM and the SE).10–15
To link these phenomena to the true or actual state-of-charge (SOC) of bulk-type SSB cells, the degree of (de)lithiation of the CAM (here, Li1+x(Ni0.6Co0.2Mn0.2)1−xO2, NCM622) during the charge/discharge process was examined in the present work. Specifically, the cathode of pelletized cells was probed both by ex situ laboratory X-ray diffraction (XRD) in reflection mode and by operando synchrotron-based XRD in transmission geometry (details in the ESI†), which also allowed detection of electrochemically inactive CAM (presence of electrically isolated particles or electrode sections).8 Of note, changes in Li content are directly reflected in lattice parameter changes, as has been shown for various NCMs in liquid electrolyte-based LIB cells.16–21 Here, operando XRD data were collected on a LIB pouch cell using the respective NCM622 CAM (contour plot of operando XRD patterns in Fig. S1, ESI†) and served as the reference standard for changes in lattice parameters upon charge and discharge.
Typically, the a-axis decreases in length in a rather linear fashion with delithiation due to contraction of the ab-plane, largely as a result of Ni oxidation (decrease in ionic radius with increasing oxidation state). In contrast, the c-axis increases at first up to x(Li) ≈ 0.5 because of electrostatic repulsion between the oxygen layers, followed by a steep decrease (contraction of the interslab distance) resulting from charge transfer between the O and Ni atoms at high SOC.16,17 This characteristic behaviour of the c-lattice parameter during cycling is depicted in Fig. 1 and was used for determining the SOC of SSB cells. The corresponding changes in a-lattice parameter and the shrinkage of unit cell volume upon charge are shown in Fig. S2 and S3 (ESI†), respectively.
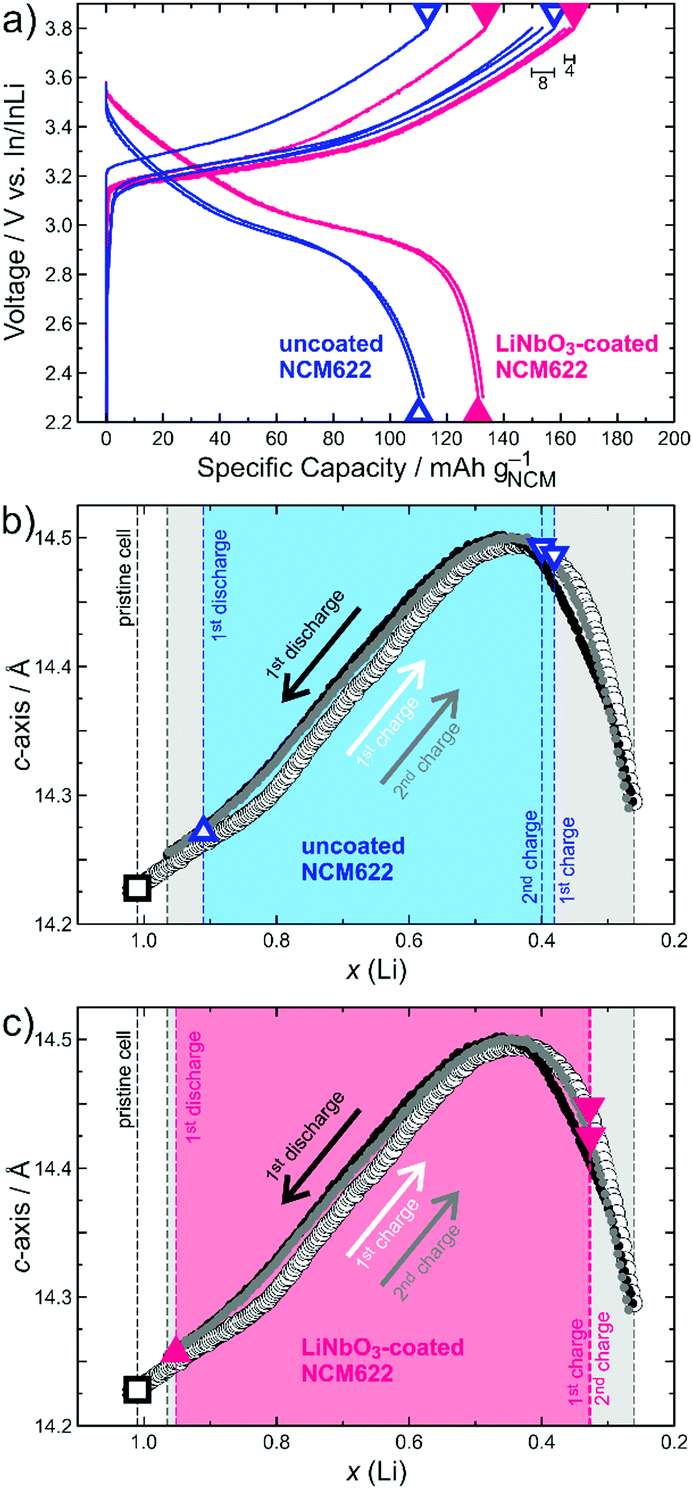 | ||
| Fig. 1 (a) Charge/discharge curves at C/10 for SSB cells using uncoated (blue) and LiNbO3-coated NCM622 (red). Different SOCs probed by means of ex situ XRD are denoted by triangles. (b and c) Changes in c-lattice parameter of NCM622 in the first charge (empty circles), first discharge (black circles) and second charge cycle (grey circles) of a liquid electrolyte-based LIB pouch cell from Rietveld analysis of operando XRD data. Note that x(Li) was calculated from the electrochemical data. Refined c-lattice parameters for (b) uncoated and (c) LiNbO3-coated NCM622 from ex situ XRD data are denoted by triangles. The black square represents the pristine (non-charged) cathode composite for comparison. Shaded areas in blue/red and grey for SSB and LIB cells, respectively, indicate the x(Li) range that can be utilized during cycling. | ||
Based on the above methodology, we have studied in this work uncoated and LiNbO3-coated NCM622 CAM in pelletized SSB cells comprising β-Li3PS4 and In metal as SE and anode, respectively. The cells were prepared by cold pressing and the cathode composite—made by milling—was composed of a 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :7 mixture (w/w) of SE and CAM. We note that carbon-free electrodes were used to keep the degradation of the lithium thiophosphate SE at a minimum.22–24 The LiNbO3 protective surface coating was applied by the sol–gel method using lithium and niobium ethoxide precursors. More details about the materials, experimental procedures and cell assembly are provided in the ESI.†
:7 mixture (w/w) of SE and CAM. We note that carbon-free electrodes were used to keep the degradation of the lithium thiophosphate SE at a minimum.22–24 The LiNbO3 protective surface coating was applied by the sol–gel method using lithium and niobium ethoxide precursors. More details about the materials, experimental procedures and cell assembly are provided in the ESI.†
For both the uncoated and LiNbO3-coated NCM622, three independent SSB cells were tested. They were assembled in a custom setup (details in the ESI†) and cycled at a rate of C/10 (1C = 180 mA g−1) in the voltage range between 2.3 and 3.8 V vs. In/InLi, corresponding to about 2.9–4.4 V vs. Li+/Li. For ex situ XRD, the different cells were stopped and disassembled after the first charge, first discharge and second charge cycle, as indicated in the voltage profiles in Fig. 1a. In charged state, an immediate voltage drop of around 100 and 200 mV for the coated and uncoated NCM622, respectively, was observed (results from relaxation measurements and galvanostatic intermittent titration technique in Fig. S4, ESI†). However, thereafter the open-circuit voltage levelled off, meaning there was virtually no further Li extraction by relaxation. The lattice parameters from Rietveld refinement analysis (details in the ESI†) are presented in Table 1 (figure of merits in Table S1, ESI†) and further denoted in Fig. 1b and c for direct comparison with the results of the liquid electrolyte-based LIB pouch cell. The Li content [x(Li)] of pristine NCM622 was determined by XRD from the cathode composite prior to cycling.
![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m). Relevant x(Li) values derived by c-lattice parameter comparison with LIB results are highlighted in bold. Refined active/inactive material fractions are also given
m). Relevant x(Li) values derived by c-lattice parameter comparison with LIB results are highlighted in bold. Refined active/inactive material fractions are also given
| NCM622 | a/Å | c/Å | x(Li) | Ratio/% | |
|---|---|---|---|---|---|
| Uncoated | 1st charge | 2.8160(1) | 14.484(1) | 0.38(2) | 93(2) |
| Inactive | 2.8672(2) | 14.221(1) | 7(2) | ||
| 1st discharge | 2.8579(1) | 14.274(1) | 0.91(2) | ||
| 2nd charge | 2.8172(1) | 14.490(1) | 0.40(2) | 91(2) | |
| Inactive | 2.8673(1) | 14.228(1) | 9(2) | ||
| Coated | 1st charge | 2.8155(1) | 14.445(1) | 0.33(2) | 92(2) |
| Inactive | 2.8669(1) | 14.220(2) | 8(2) | ||
| 1st discharge | 2.8614(1) | 14.259(1) | 0.95(2) | ||
| 2nd charge | 2.8153(2) | 14.422(2) | 0.33(2) | 96(3) | |
| Inactive | 2.8662(2) | 14.232(2) | 4(3) | ||
| Pristine | 2.8687(1) | 14.228(1) | 1.01(2) | ||
From the charge/discharge curves in Fig. 1a, it can be seen that the SSB cells comprising the LiNbO3-coated NCM622 delivered higher specific capacities (e.g. qch = 165 mA h g−1 and qdis = 133 mA h g−1 in the initial cycle), with an improved Coulombic efficiency of 81% vs. 72% when compared to the uncoated CAM (qch = 154 mA h g−1 and qdis = 111 mA h g−1). The same trend is also apparent from the capacity retention data over 60 cycles in Fig. S5 (ESI†). Notably, the different SSB cells tested showed only minor variations in the initial specific charge capacity (8 and 4 mA h g−1 for the uncoated and LiNbO3-coated NCM622, respectively), as indicated in Fig. 1a. This, in turn, allowed for reasonable determination of x(Li) after charge and discharge via facile comparison of refined c-lattice parameters (see shaded areas in Fig. 1b and c). Likewise, SOC determination based on the changes in a-lattice parameter during cycling is possible and, as is evident from Fig. S2 (ESI†), there are only slight deviations in x(Li) between a and c.
Overall, Fig. 1 and Table 1 provide clear evidence that the actual amount of Li that can be cycled in SSB cells is higher for the LiNbO3-coated NCM622 [x(Li) ≈ 0.62, compared to about 0.51 for the uncoated CAM]. This result is not only due to the higher degree of delithiation achieved in the initial charge cycle, but also because of improved lithiation (re-insertion of Li into the host lattice) during the subsequent discharge process. The data in Fig. 1a suggest that part of the reason is the lower charge-transfer resistance at the CAM/SE interface after surface coating. For the uncoated NCM622, the resistance build-up due to side reactions (cathode SEI formation), especially during the initial charge cycle, and/or partial contact loss caused by shrinkage of the CAM particles (mechanical degradation) lead to significantly higher overvoltage, which also helps to explain the relatively low first cycle Coulombic efficiency. Note that x(Li) or, in other words, the SOC achieved in the first and second charge cycles remains virtually unaltered for the LiNbO3-coated NCM622, while it slightly changes for the uncoated CAM.
Ex situ XRD analysis suggests that the initial capacity decay is not related to significant contact loss of CAM. Detachment of SE from the NCM622 secondary particles may occur because of volume change effects during cycling operation.7,25 A complete loss of contact would lead to the appearance of two NCM phase fractions after discharge, namely, a major one representing the electrochemically connected and lithiated NCM622 and a minor one for the detached (electrically isolated) and still delithiated NCM622. However, for both the uncoated and LiNbO3-coated NCM622, XRD confirmed the presence of only a single phase. As mentioned previously, differently charged or discharged species would result in splitting/broadening of NCM622 reflections (especially noticeable for the 003 reflection). The 003 reflection of the layered CAM for all ex situ XRD patterns collected in this work is shown in Fig. 2 (Rietveld plots of XRD data in the 2θ range of 10–90° in Fig. S6, ESI†). A single reflection is visible after the first discharge cycle and at a larger angle for the LiNbO3-coated NCM622, indicating an overall higher degree of lithiation (smaller c-lattice parameter, with sin(θ003) = 3/2·λ/c). In contrast, after the first and second charge cycles, at least two NCM622 species of different x(Li) are observed for both the uncoated and LiNbO3-coated NCM622. The intense diffraction at a relatively lower 2θ angle represents the delithiated (active) CAM fraction (c-lattice parameters denoted in Fig. 1), while the weaker reflection at a larger angle represents an electrochemically inactive fraction. The refined lattice parameters match well with those for the pristine cathode composite (Table 1), indicating that no substantial amount of lithium was extracted during the charge process. Of note, we only considered two NCM622 fractions (active/inactive) in the Rietveld analysis, which does not necessarily represent the true picture. As recently demonstrated by X-ray absorption spectroscopy imaging of SSB cells using slurry-coated cathodes, several electrode fractions of different SOC may be present during cycling.26 As can be seen in Fig. 2, the 003 reflection of charged material exhibits asymmetric broadening (toward higher 2θ values), especially for the LiNbO3-coated NCM622 after the second charge cycle, thus hinting at the presence of at least one additional CAM fraction. Interestingly, the refined active/inactive material fractions (Table 1 and Fig. 2) are similar for both the uncoated and LiNbO3-coated NCM622. Hence, the inactive CAM seems to be present right from the beginning and, probably, is due to some inhomogeneity in the cathode composites used. In our previous work, we have shown that the CAM secondary particle size is decisive for ensuring sufficient electronic pathways in carbon-free composite electrodes.8
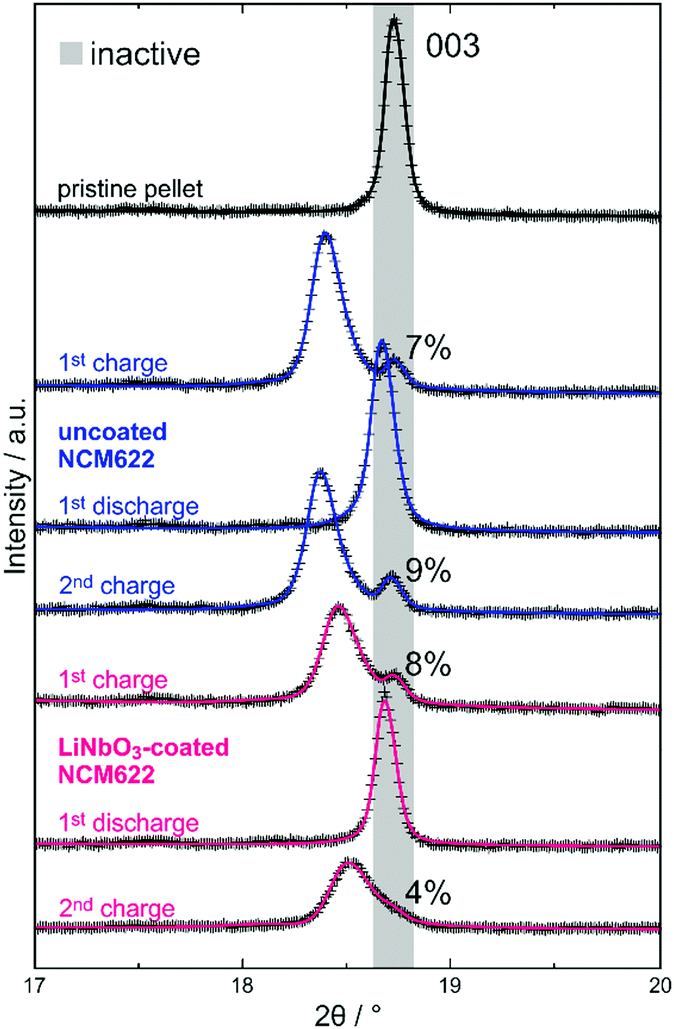 | ||
| Fig. 2 Rietveld plots of ex situ XRD data (Cu-Kα) in the 2θ range of the 003 reflection for uncoated (blue) and LiNbO3-coated NCM622 (red) after the first charge, first discharge and second charge cycle. The pattern for pristine (non-charged) cathode composite is also displayed. Observed and calculated patterns are shown as crosses and solid lines, respectively. The shaded area represents the characteristic 2θ range for pristine (non-charged), fully discharged and inactive NCM622 (in charged cells; fraction given in percent). | ||
From the refined active CAM fraction and estimated SOC [x(Li)] from the ex situ and operando XRD results, the specific capacity (based on the total mass of NCM622) of the initial charge cycle can be calculated and eventually compared to that measured by galvanostatic cycling. In doing so, qch = 162 and 173 mA h g−1 were obtained for the uncoated and LiNbO3-coated NCM622, respectively (details in the ESI†). The difference of ∼10 mA h g−1 is in good agreement with that observed for the experimental qch values (154 and 165 mA h g−1). The fact that the calculated capacities are larger than the electrochemically measured ones may be due to the simplified assumption of the presence of only two NCM phases during charge. However, non-negligible errors occur in both estimation of the active CAM fraction and determination of x(Li), making differences in qch between the calculated and experimental data reasonable (details in the ESI†). Note that similar calculations for the initial discharge and second charge cycles are inappropriate, as they are strongly affected by cumulative errors. However, our approach clearly demonstrates that data from galvanostatic cycling experiments do not necessarily reflect the true lithium content of CAM/actual SOC of bulk-type SSB cells, which can be determined by XRD, though. If the entire CAM had been active, initial specific charge capacities of 174 and 188 mA h g−1 would have been achieved for the uncoated and LiNbO3-coated NCM622, respectively, emphasising the importance of considering the presence of inactive material in such cells. Also, ex situ XRD revealed similar SOCs for the initial and second charge cycles. Hence, the large irreversibility in the first cycle can indeed be attributed to side reactions between the NCM622 and β-Li3PS4 and/or incomplete re-lithiation upon discharge due to hindered kinetics (partial mechanical contact loss).
The reliability of the comparative approach presented here is based on the conformity of the electrochemical behaviour of solid and liquid electrolyte cells when using the same CAM. However, studying SSB cells ex situ leads to an additional error contribution (potentially lower for slurry-coated cathodes than pelletized electrodes). Consequently, it may seem sensible to conduct operando XRD measurements on single cells, which should also allow thoroughly examining the behaviour of SE during cycling. Such preliminary experiments were carried out on bulk-type model cells at the light source PETRA III at DESY using a custom setup, enabling synchrotron-based XRD in transmission geometry (high-flux hard X-rays guaranteed sufficient penetration through the pelletized cell as well as high signal-to-noise ratio and good time resolution). However, for experimental reasons, so far, it was only possible to study cells comprising argyrodite-type Li6PS5Cl as SE, which exhibits a higher ionic conductivity than β-Li3PS4 (details in the ESI†). In general, similar observations regarding the presence of differently charged particles and changes in lattice parameters of NCM622 upon charge/discharge were made (contour plot of operando XRD patterns in Fig. S7, ESI†). In addition, operando XRD revealed that the lattice parameters of Li6PS5Cl remain virtually unaltered during cycling, which is due in part to the strong contribution from the separator layer, yet this needs further study.
In summary, we have shown that XRD is a viable method to indirectly determine the actual state-of-charge of SSBs (the amount of cyclable Li in such cells cannot be calculated in a reliable way from electrochemical testing data only). In addition, it lends itself to uncover the presence of electrochemically inactive electrode material, which may help to develop advanced cathode composites for next-generation SSB applications.
The authors acknowledge the German Ministry of Education and Research (BMBF) for funding within the project ARTEMYS (03XP0114J). This work is part of the projects being funded within the BASF International Network for Batteries and Electrochemistry. Parts of this research were carried out at the light source PETRA III at DESY, a member of the Helmholtz Association (HGF). We thank Martin Tolkiehn for assistance in using beamline P24 (EH2).
Conflicts of interest
The authors declare no competing financial interest.References
- Z. Gao, H. Sun, L. Fu, F. Ye, Y. Zhang, W. Luo and Y. Huang, Adv. Mater., 2018, 30, 1705702 CrossRef PubMed.
- J. Janek and W. G. Zeier, Nat. Energy, 2016, 1, 16141 CrossRef.
- Y. S. Jung, D. Y. Oh, Y. J. Nam and K. H. Park, Isr. J. Chem., 2015, 55, 472 CrossRef CAS.
- J. Kasemchainan and P. G. Bruce, Johnson Matthey Technol. Rev., 2018, 62, 177 CrossRef CAS.
- J. Schnell, T. Günther, T. Knoche, C. Vieider, L. Köhler, A. Just, M. Keller, S. Passerini and G. Reinhart, J. Power Sources, 2018, 382, 160 CrossRef CAS.
- K. H. Park, Q. Bai, D. H. Kim, D. Y. Oh, Y. Zhu, Y. Mo and Y. S. Jung, Adv. Energy Mater., 2018, 8, 1800035 CrossRef.
- R. Koerver, I. Aygün, T. Leichtweiß, C. Dietrich, W. Zhang, J. O. Binder, P. Hartmann, W. G. Zeier and J. Janek, Chem. Mater., 2017, 29, 5574 CrossRef CAS.
- F. Strauss, T. Bartsch, L. de Biasi, A.-Y. Kim, J. Janek, P. Hartmann and T. Brezesinski, ACS Energy Lett., 2018, 3, 992 CrossRef CAS.
- Y. J. Nam, D. Y. Oh, S. H. Jung and Y. S. Yung, J. Power Sources, 2018, 375, 93 CrossRef CAS.
- J. Haruyama, K. Sodeyama, L. Y. Han, K. Takada and Y. Tateyama, Chem. Mater., 2014, 26, 4248 CrossRef CAS.
- N. Ohta, K. Takada, L. Q. Zhang, R. Z. Ma, M. Osada and T. Sasaki, Adv. Mater., 2006, 18, 2226 CrossRef CAS.
- N. Ohta, K. Takada, I. Sakaguchi, L. Q. Zhang, R. Z. Ma, K. Fukuda, M. Osada and T. Sasaki, Electrochem. Commun., 2007, 9, 1486 CrossRef CAS.
- C. Wang, L. Chen, H. Zhang, Y. Yang, F. Wang, F. Yin and G. Yang, Electrochim. Acta, 2014, 119, 236 CrossRef CAS.
- P. Heitjans, M. Masoud, A. Feldhoff and M. Wilkening, Faraday Discuss., 2007, 134, 67 RSC.
- D. H. Kim, D. Y. Oh, K. H. Park, Y. E. Choi, Y. J. Nam, H. A. Lee, S.-M. Lee and Y. S. Jung, Nano Lett., 2017, 17, 3013 CrossRef CAS PubMed.
- L. de Biasi, A. O. Kondrakov, H. Geßwein, T. Brezesinski, P. Hartmann and J. Janek, J. Phys. Chem. C, 2017, 121, 26163 CrossRef.
- A. O. Kondrakov, H. Geßwein, K. Galdina, L. de Biasi, V. Meded, E. O. Filatova, G. Schumacher, W. Wenzel, P. Hartmann, T. Brezesinski and J. Janek, J. Phys. Chem. C, 2017, 121, 24381 CrossRef CAS.
- A. O. Kondrakov, A. Schmidt, J. Xu, H. Geßwein, R. Mönig, P. Hartmann, H. Sommer, T. Brezesinski and J. Janek, J. Phys. Chem. C, 2017, 121, 3286 CrossRef CAS.
- H.-J. Noh, S. Youn, C. S. Yoon and Y.-K. Sun, J. Power Sources, 2013, 233, 121 CrossRef CAS.
- H.-H. Sun and A. Manthiram, Chem. Mater., 2017, 29, 8486 CrossRef CAS.
- H.-H. Ryu, K.-J. Park, C. S. Yoon and Y.-K. Sun, Chem. Mater., 2018, 30, 1155 CrossRef CAS.
- W. Zhang, T. Leichtweiß, S. P. Culver, R. Koerver, D. Das, D. A. Weber, W. G. Zeier and J. Janek, ACS Appl. Mater. Interfaces, 2017, 9, 35888 CrossRef CAS PubMed.
- T. Hakari, M. Deguchi, K. Mitsuhara, T. Ohta, K. Saito, Y. Orikasa, Y. Uchimoto, Y. Kowada, A. Hayashi and M. Tatsumisago, Chem. Mater., 2017, 29, 4768 CrossRef CAS.
- R. Koerver, F. Walther, I. Aygün, J. Sann, C. Dietrich, W. G. Zeier and J. Janek, J. Mater. Chem. A, 2017, 5, 22750 RSC.
- R. Koerver, W. Zhang, L. de Biasi, S. Schweidler, A. O. Kondrakov, S. Kolling, T. Brezesinski, P. Hartmann, W. G. Zeier and J. Janek, Energy Environ. Sci., 2018, 11, 2142 RSC.
- K. Chen, S. Shinjo, A. Sakuda, K. Yamamoto, T. Uchiyama, K. Kuratani, T. Takeuchi, Y. Orikasa, A. Hayashi, M. Tatsumisago, Y. Kimura, T. Nakamura, K. Amezawa and Y. Uchimoto, J. Phys. Chem. C, 2019, 123, 3292 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Materials and experimental procedures; contour plots of operando XRD patterns for liquid and solid electrolyte-based cells; evolution of c- and a-lattice parameters and unit cell volume with (de)lithiation; results from relaxation and GITT measurements; long-term cycling data; and Rietveld plots of ex situ XRD data. See DOI: 10.1039/c9cc04453a |
| This journal is © The Royal Society of Chemistry 2019 |