
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNew insights into solvent-induced structural changes of 13C labelled metal–organic frameworks by solid state NMR†
Marcus
Rauche‡
a,
Sebastian
Ehrling‡
b,
Simon
Krause§
 b,
Irena
Senkovska
b,
Stefan
Kaskel
b and
Eike
Brunner
*a
b,
Irena
Senkovska
b,
Stefan
Kaskel
b and
Eike
Brunner
*a
aChair of Bioanalytical Chemistry, TU Dresden, D-01062 Dresden, Germany. E-mail: eike.brunner@tu-dresden.de
bChair of Inorganic Chemistry I, TU Dresden, D-01062 Dresden, Germany
First published on 8th July 2019
Abstract
Selective 13C-labelling of carboxylate carbons in the linker molecules of flexible metal–organic frameworks (MOFs) makes solid-state NMR spectroscopy very powerful to investigate solvent-induced local structural changes as demonstrated by 13C and 1H NMR spectroscopy on the pillared layer MOF DUT-8(Ni). Selective identification of polar solvent–node interactions becomes feasible.
Metal–organic frameworks (MOFs) are versatile materials of exceptional porosity1,2 and promising for applications in gas and liquid phase adsorption, gas storage, separation processes, catalysis, and sensing.3–6 The framework crystal structure is usually determined by X-ray or neutron diffraction. In contrast, defects, linker disorder, as well as disordered guest molecules and their interactions with the framework are often difficult to study by diffraction techniques. Recently, Yaghi and co-workers demonstrated the impact of disordered solvent molecules on crystallographic experiments illustrating the need for extended analysis of the solvents present within the pores of MOFs.7 Solvents play a key role for MOFs. They are important for cluster formation and crystallisation in solvothermal syntheses,8 for solvent exchange to remove side products and unreacted starting materials, solvent removal to obtain guest-free porosity,9,10 and finally for filling the pores with fluids11,12 in various applications. In most cases, solvents have only minor influence upon the structure of the framework (rigid MOFs). However, flexible MOFs can undergo massive changes of structure and unit cell volume in response to the adsorption of certain guest molecules.13 In some cases, specific interactions of solvent molecules with flexible frameworks are found to dictate the structural response.14–16 Selective recognition of solvents with different polarity and chemical constitution was recently reported for DUT-8(Ni) with potential sensing applications.17 However, selective adsorption and guest recognition in flexible MOFs are not fully understood yet. Further in-depth investigations are necessary.
NMR is widely used to study MOFs and their host–guest interactions with adsorbed species by probing either the host or the guest molecules.18–31 Sensitivity is often a major limitation of NMR spectroscopy, especially for nuclei with low natural abundance like 13C. Isotope enrichment is then highly desirable. In the present contribution, we describe the application of solid-state NMR to study the interaction of various organic solvent molecules with the flexible MOF DUT-8(Ni)32 (Ni2(2,6-ndc)2(dabco) (2,6ndc = 2,6-naphthalene-dicarboxylate, dabco = 1,4-diazabicyclo[2.2.2]octane)) to analyse structural changes and underlying, potentially selective host–guest interactions. Solvothermally synthetized DUT-8(Ni) crystals with sizes beyond 1 μm (compound 1) undergo a pronounced structural transformation during desolvation. The framework switches from an open pore (op) to a closed pore (cp) phase. The structure is re-opened by adsorption of appropriate gases or liquids, resulting in a “gate-opening behaviour”. The flexibility of 1 was characterized from a fundamental point of view in several studies.22,32–40 In contrast, DUT-8(Ni) with crystal sizes below 1 μm, further denoted as compound 2, is rigid and remains in a metastable open pore phase even after guest removal (Fig. S1–S3, ESI†).22,36
13C cross polarisation magic angle spinning (CP MAS) NMR spectra of DUT-8(Ni) without isotope enrichment exhibit low intensity especially for non-protonated linker carboxylate carbon atoms.22,33 Thus, the experiments are very time-consuming and the resulting spectra of low quality. To enhance the sensitivity, carbon atoms of the carboxyl groups of 2,6-H2ndc were selectively labelled with 13C by lithiation of 2,6-dibromonaphthalene and subsequent carboxylation with 13C-enriched CO2 (Scheme 1, for details see Section 4 of ESI,† Fig. S4–S6). Because many MOFs are based on linkers containing carboxylic acids,41 this synthetic approach can be widely applied to provide a strong and selective sensitivity enhancement for solid-state 13C NMR experiments.
 | ||
| Scheme 1 Reaction scheme of the 13C-labelling of 2,6-dibromonaphtalene by lithiation and subsequent carboxylation with 13C-enriched CO2. | ||
Fig. 1 shows the 13C (1H) CP MAS NMR spectra of 2,6-H2ndc with selectively 13C-enriched carboxylic acid functionalities and of as made flexible DUT-8(Ni) solvothermally synthesized in N,N-dimethylformamide (DMF) (compound 1). Remarkably, linker incorporation into the MOF lattice causes a huge increase of the isotropic 13C NMR chemical shift from 173 ppm to 269 ppm and of the static linewidth as reflected by the broad spinning sideband pattern. Second moments, M2, are frequently used measures for line broadening. They can be calculated from the spinning sideband intensities (Fig. S8 and S9, ESI†). An increase of M2 from 2 × 109 s−2 for the pure linker to 2 × 1010 s−2 for DUT-8(Ni) occurs at a field corresponding to 800 MHz 1H resonance frequency. This may be due to two different reasons: (i) an increase of the chemical shift anisotropy due to the coordination of the carboxylate groups to the Ni2 cluster (Fig. 1) and/or (ii) a paramagnetic broadening due to the neighbouring nickel atoms. For both interactions, M2 should exhibit a field dependence proportional to the square of the static magnetic field, B0, of the spectrometer as is indeed observed (Fig. S9, ESI†).42–45 In contrast, dipolar interactions can be excluded as dominating source of line broadening because the second moment should then be independent of B0. The remarkably high isotropic 13C NMR chemical shift of 269 ppm points towards a possible paramagnetic shift of the signal due the neighbouring Ni2 sites. Earlier magnetisation measurements32 indeed show a paramagnetic susceptibility which is, however, lower than for typical paramagnetic compounds (ESI,† page 11).
 | ||
| Fig. 1 13C CP MAS NMR spectra of 2,6-H2ndc with selectively 13C-labelled carboxylate groups and of 1 containing 2,6-ndc with selectively 13C-labelled carboxylates measured in DMF. Complete signal assignment is provided in Fig. S7, ESI.† The measurements were carried out at a magnetic field corresponding to 800 MHz 1H resonance frequency. | ||
Furthermore, the removal of the solvent from compound 1 leads to structure closing accompanied by an increase of the magnetic susceptibility by a factor of about four.32 If paramagnetism would have a major effect upon the isotropic chemical shift, solvent removal should result in a further chemical shift increase. The opposite is true, solvent removal reduces the chemical shift to 229 ppm for the carboxylates. Paramagnetic effects can thus not be the dominating source of the high isotropic chemical shift in the as made samples. This conclusion is supported by the absence of EPR signals,35,40 calculations46 predicting an antiferromagnetic coupling in the Ni2 cluster, and the temperature dependence of the carboxylate 13C NMR signal (Fig. S10, ESI†).
Comparison of the spectra for the flexible compound 1 and the rigid compound 2 reveals that the 13C NMR spectra of both materials exhibit very similar chemical shifts. This is true for the as made and the desolvated state (Fig. 2). The fact, that desolvated 2 (op form) exhibits almost the same chemical shift as desolvated 1 (cp form) reveals that the solvent-induced structural transition of compound 1 does not significantly influence the isotropic chemical shift of linker carboxylate groups. Furthermore, the residual linewidth of the 13C CP MAS NMR signal of the linker carboxylates is significantly larger for as made compound 2 compared to 1 (Fig. S11, ESI†). This can be ascribed to a higher degree of linker disorder for compound 2 in agreement with previous observations and may explain the fact that compound 2 is non-flexible, i.e., unable to undergo the structural transition observed in 1 after solvent removal.22
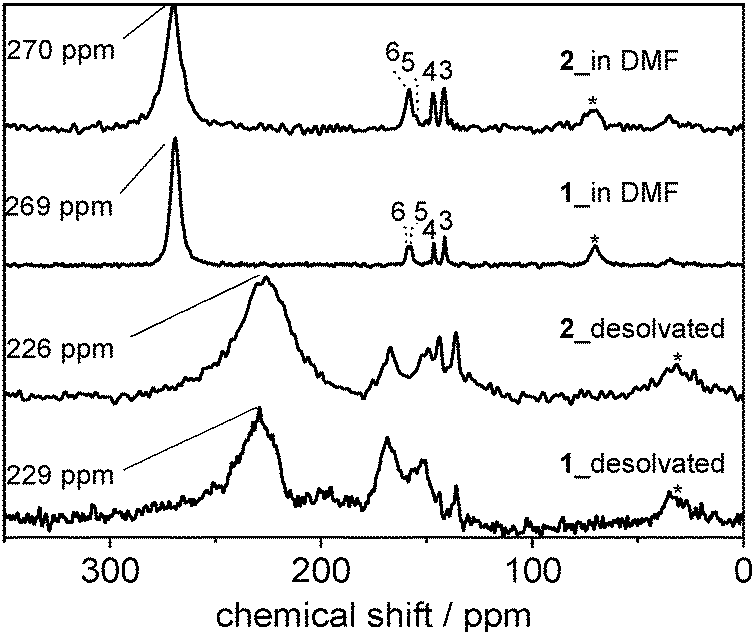 | ||
| Fig. 2 13C CP MAS NMR spectra of 1 and 2 containing 2,6-ndc with selectively 13C-labelled carboxylates in the solvated (DMF) and solvent free (desolvated) state. Signal assignment is provided in Fig. S7, ESI.† | ||
To further elucidate the reason for the large isotropic chemical shift of the as made, DMF-containing samples and its decrease during solvent removal, we systematically varied the solvent. A clear correlation between solvent polarity and isotropic 13C chemical shift of the carboxylates is observed (Fig. 3, Table S4 and Fig. S12, ESI†). Note that the open pore state of DUT-8(Ni) filled with nonpolar solvents like n-alkanes exhibits practically the same isotropic chemical shift of ca. 230 ppm as the closed, solvent-free sample (Fig. 3 and Fig. S12, ESI†). More polar solvents like DMF give rise to a chemical shift change up to ca. 40 ppm relative to the latter value.
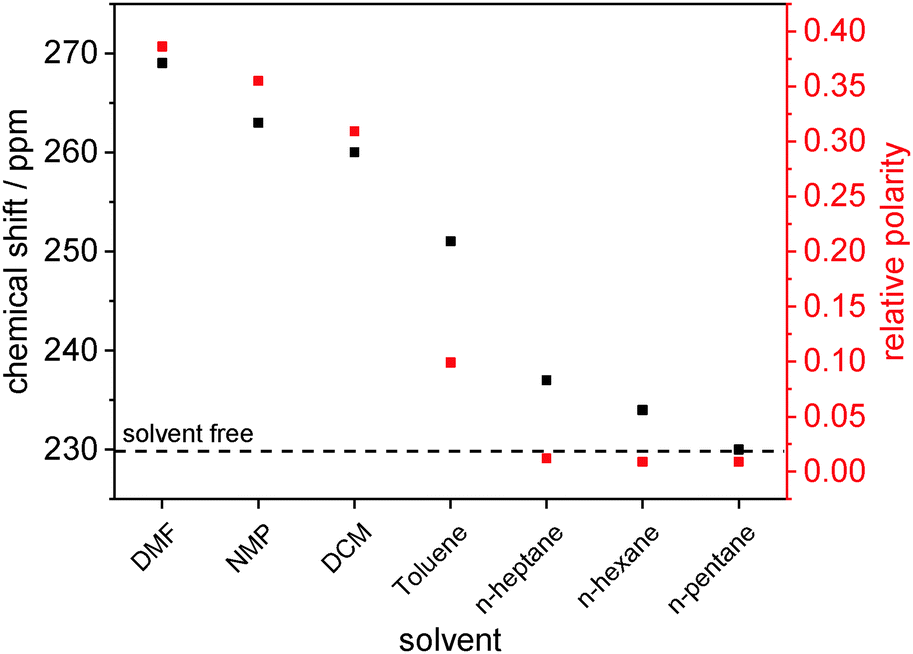 | ||
| Fig. 3 13C NMR chemical shift of the carboxylate groups measured for 1 containing different solvents in the pores (NMP: N-methyl-2-pyrrolidone; DMF: N,N-dimethylformamide; DCM: dichloromethane). The relative polarity of the used solvents (cf. Table S4, ESI†) is shown in red. | ||
Potentially, this indicates a specific interaction of the solvent molecules with the paddle wheel units. This hypothesis is further corroborated by 1H MAS NMR studies (cf.Fig. 4 and Section 7, ESI† with Fig. S13–S15). A broad signal centred at about 8 ppm occurs for the closed, solvent-free form of 1 due to 1H nuclei located at the aromatic linkers. Moreover, the 1H nuclei of the dabco molecules also contribute to this signal. It is remarkable that the 1H signals of CH2 groups in dabco molecules occur at only ca. 3.5 ppm in solution. Solvating 1 with nonpolar solvents like n-heptane results in a decent shift of the broad signal to about 9–10 ppm. However, an additional signal at ca. 12–13 ppm occurs for the polar solvents DMF, N-methyl-2-pyrrolidone (NMP) and dichloromethane (DCM). Using samples containing deuterated linker and/or dabco molecules (compounds 3, 4, 5, for details see Section S1 and Table S1, ESI†), we could assign this additional signal to CH2 groups of dabco molecules (see ESI,† Section 7 and Fig. S13). This assignment is confirmed by the 2D 1H-13C-HETCOR spectrum, which shows a cross polarisation between the CH2 carbon atom of dabco at ca. 35 ppm and 1H nuclei at ca. 13 ppm for DMF and 10 ppm for n-heptane loaded samples at short mixing times (Fig. S16, ESI†). Relatively high 1H NMR chemical shifts are often considered as indication for hydrogen bond formation.47 This unexpected observation points towards a specific interaction of polar solvent molecules with the pillaring dabco molecules in DUT-8(Ni). Previous observations suggesting the formation of specific adsorption complexes in the neighbourhood of metal sites in MOFs support this idea.48,49 Former studies on DUT-8(Ni) showed distinct differences in the adsorption behaviour of aprotic polar and protic polar solvents like alcohols.17 To investigate the influence of solvent molecules that can act as donor and acceptor (amphiprotic behaviour), we performed a series of experiments with various alcohols. Surprisingly, we could not find a direct correlation between the polarity of the alcohols and the chemical shift in the 13C spectra (Fig. S17, ESI†). The chemical shift decreases from 249 ppm (ethanol) to 242 ppm (propanol) indicating a different interaction with the framework. The chemical shift then increases with further increasing size of the alcohol up to 254 ppm for octanol – well below the high shift value of ca. 270 ppm observed, e.g., for DMF (Fig. 3). Alcohols are amphiprotic, but act more as proton donor than acceptor sites resulting in weaker interactions with the framework. This agrees with previous observations showing that adsorption of alcohols cannot trigger the structural transformation of the desolvated flexible compound 1.17 Further investigations are necessary in order to clarify the special behaviour of alcohols.
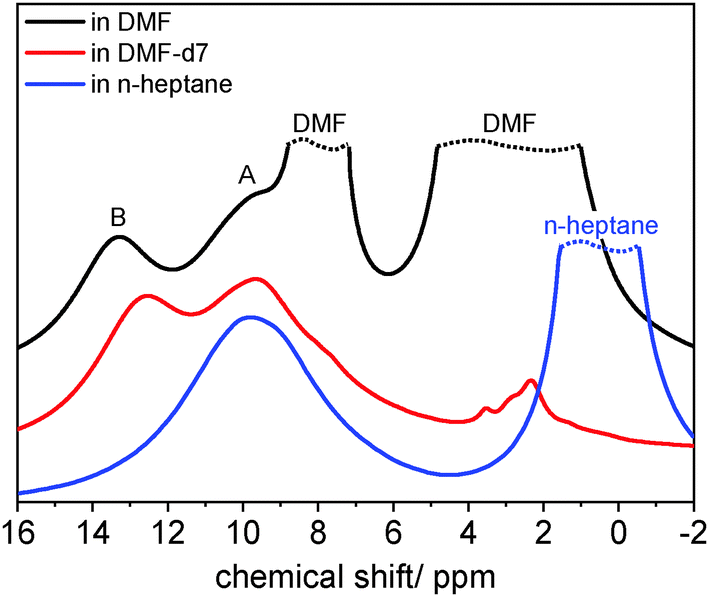 | ||
| Fig. 4 1H MAS NMR spectra of 1 loaded with DMF and n-heptane. The exchange of DMF by deuterated DMF reveals the two major signals, A and B, belonging to the MOF structure in samples loaded with polar solvents. Minor signals at 2–4 ppm for the sample exchanged with DMF-d7 are due to residual amounts of protonated DMF. | ||
In summary, we report selective 13C-labelling of the carboxylate group of the 2,6-H2ndc linker used in DUT-8(Ni). This technique provides excellent sensitivity for selectively investigating local structural changes of the nodes of flexible MOFs. Solvent polarity exerts a pronounced influence upon the chemical shift. Nonpolar solvents exhibit weaker interactions with the nodes of the framework. Polar solvents strongly interact with the framework clusters and give rise to substantial shifts of the carboxylate 13C NMR signals. Furthermore, the protons of dabco exhibit a surprisingly high chemical shift depending on solvent polarity, possibly by hydrogen bond formation involving dabco molecules. In addition, the fact that large crystallites of DUT-8(Ni) are flexible in contrast to small crystallites does not originate from different solid-fluid interactions as could be derived from the identity of the spectroscopic signatures for both structural variants. Only one difference between the flexible and rigid variant was detected by 13C NMR spectroscopy: the residual line width of the carboxylate 13C CP MAS NMR signal of the rigid compound 2 is considerably higher than for the flexible compound 1. Based on the isotope-labelling scheme proposed here, future spectroscopic investigations will enable the in-depth analysis of site-specific host–guest interactions and complexes formed by the adsorption of polar solvents in MOFs. This could open up prospects for selectively tuning the adsorption properties by rational materials design towards enhanced selectivity.
Financial support from the Deutsche Forschungsgemeinschaft (FOR 2433 “MOF-Switches”) is gratefully acknowledged.
Conflicts of interest
There are no conflicts to declare.Notes and references
- O. K. Farha, I. Eryazici, N. C. Jeong, B. G. Hauser, C. E. Wilmer, A. A. Sarjeant, R. Q. Snurr, S. T. Nguyen, A. Ö. Yazaydın and J. T. Hupp, J. Am. Chem. Soc., 2012, 134, 15016 CrossRef CAS.
- I. M. Hönicke, I. Senkovska, V. Bon, I. A. Baburin, N. Bönisch, S. Raschke, J. D. Evans and S. Kaskel, Angew. Chem., Int. Ed., 2018, 57, 13780 CrossRef.
- A. Schneemann, V. Bon, I. Schwedler, I. Senkovska, S. Kaskel and R. A. Fischer, Chem. Soc. Rev., 2014, 43, 6062 RSC.
- S. Kaskel, The Chemistry of Metal-Organic Frameworks, 2 Volume Set: Synthesis, Characterization, and Applications, John Wiley & Sons, 2016 Search PubMed.
- D. Farrusseng, Metal-organic frameworks: applications from catalysis to gas storage, John Wiley & Sons, 2011 Search PubMed.
- Z. Chang, D. H. Yang, J. Xu, T. L. Hu and X. H. Bu, Adv. Mater., 2015, 27, 5432 CrossRef CAS.
- S. Lee, H.-B. Bürgi, S. A. Alshmimri and O. M. Yaghi, J. Am. Chem. Soc., 2018, 140, 8958 CrossRef CAS.
- B. Seoane, A. Dikhtiarenko, A. Mayoral, C. Tellez, J. Coronas, F. Kapteijn and J. Gascon, CrystEngComm, 2015, 17, 1693 RSC.
- R. A. Dodson, A. G. Wong-Foy and A. J. Matzger, Chem. Mater., 2018, 30, 6559 CrossRef CAS.
- J. Ma, A. P. Kalenak, A. G. Wong-Foy and A. J. Matzger, Angew. Chem., Int. Ed., 2017, 56, 14618 CrossRef CAS.
- J. Yu and P. B. Balbuena, J. Phys. Chem. C, 2013, 117, 3383 CrossRef CAS.
- X. Jiang, H.-B. Duan, S. I. Khan and M. A. Garcia-Garibay, ACS Cent. Sci., 2016, 2, 608 CrossRef CAS.
- S. Horike, S. Shimomura and S. Kitagawa, Nat. Chem., 2009, 1, 695 CrossRef CAS.
- D. N. Dybtsev, H. Chun and K. Kim, Angew. Chem., Int. Ed., 2004, 43, 5033 CrossRef CAS.
- Y. Sakata, S. Furukawa, M. Kondo, K. Hirai, N. Horike, Y. Takashima, H. Uehara, N. Louvain, M. Meilikhov and T. Tsuruoka, Science, 2013, 339, 193 CrossRef CAS.
- M. Shivanna, Q.-Y. Yang, A. Bajpai, S. Sen, N. Hosono, S. Kusaka, T. Pham, K. A. Forrest, B. Space, K. Susumu and M. J. Zaworotko, Sci. Adv., 2018, 4, eaaq1636 CrossRef.
- N. Kavoosi, T. Savchenko, I. Senkovska, M. Maliuta, V. Bon, A. Eychmüller and S. Kaskel, Microporous Mesoporous Mater., 2018, 271, 169 CrossRef CAS.
- H. Hoffmann, M. Debowski, P. Müller, S. Paasch, I. Senkovska, S. Kaskel and E. Brunner, Materials, 2012, 5, 2537 CrossRef CAS.
- A. Sutrisno and Y. Huang, Solid State Nucl. Magn. Reson., 2013, 49, 1 CrossRef.
- V. J. Witherspoon, J. Xu and J. A. Reimer, Chem. Rev., 2018, 118, 10033 CrossRef CAS.
- Y. A. Wong, V. Martins, B. E. Lucier and Y. Huang, Chem. – Eur. J., 2019, 25, 1848 CrossRef CAS.
- N. Kavoosi, V. Bon, I. Senkovska, S. Krause, C. Atzori, F. Bonino, J. Pallmann, S. Paasch, E. Brunner and S. Kaskel, Dalton Trans., 2017, 46, 4685 RSC.
- C. Lieder, S. Opelt, M. Dyballa, H. Henning, E. Klemm and M. Hunger, J. Phys. Chem. C, 2010, 114, 16596 CrossRef CAS.
- J. Schaber, S. Krause, S. Paasch, I. Senkovska, V. Bon, D. M. Többens, D. Wallacher, S. Kaskel and E. Brunner, J. Phys. Chem. C, 2017, 121, 5195 CrossRef CAS.
- G. P. Bignami, Z. H. Davis, D. M. Dawson, S. A. Morris, S. E. Russell, D. McKay, R. E. Parke, D. Iuga, R. E. Morris and S. E. Ashbrook, Chem. Sci., 2018, 9, 850 RSC.
- J. D. Martell, L. B. Porter-Zasada, A. C. Forse, R. L. Siegelman, M. I. Gonzalez, J. Oktawiec, T. Runčevski, J. Xu, M. Srebro-Hooper and P. J. Milner, J. Am. Chem. Soc., 2017, 139, 16000 CrossRef CAS PubMed.
- S. Dey, A. Bhunia, H. Breitzke, P. B. Groszewicz, G. Buntkowsky and C. Janiak, J. Mater. Chem. A, 2017, 5, 3609 RSC.
- R. M. Marti, J. D. Howe, C. R. Morelock, M. S. Conradi, K. S. Walton, D. S. Sholl and S. E. Hayes, J. Phys. Chem. C, 2017, 121, 25778 CrossRef CAS.
- T. W. Kemnitzer, C. B. Tschense, T. Wittmann, E. A. Rössler and J. Senker, Langmuir, 2018, 34, 12538 CrossRef CAS PubMed.
- T. Wittmann, A. Mondal, C. B. Tschense, J. J. Wittmann, O. Klimm, R. Siegel, B. Corzilius, B. Weber, M. Kaupp and J. Senker, J. Am. Chem. Soc., 2018, 140, 2135 CrossRef CAS.
- T. Wittmann, C. B. Tschense, L. Zappe, C. Koschnick, R. Siegel, R. Staeglich, B. V. Lotsch and J. Senker, J. Mater. Chem. A, 2019, 7, 10379 RSC.
- N. Klein, C. Herzog, M. Sabo, I. Senkovska, J. Getzschmann, S. Paasch, M. R. Lohe, E. Brunner and S. Kaskel, Phys. Chem. Chem. Phys., 2010, 12, 11778 RSC.
- N. Klein, H. C. Hoffmann, A. Cadiau, J. Getzschmann, M. R. Lohe, S. Paasch, T. Heydenreich, K. Adil, I. Senkovska and E. Brunner, J. Mater. Chem., 2012, 22, 10303 RSC.
- A. Krylov, A. Vtyurin, P. Petkov, I. Senkovska, M. Maliuta, V. Bon, T. Heine, S. Kaskel and E. Slyusareva, Phys. Chem. Chem. Phys., 2017, 19, 32099 RSC.
- M. Mendt, F. Gutt, N. Kavoosi, V. Bon, I. Senkovska, S. Kaskel and A. Pöppl, J. Phys. Chem. C, 2016, 120, 14246 CrossRef CAS.
- H. Miura, V. Bon, I. Senkovska, S. Ehrling, S. Watanabe, M. Ohba and S. Kaskel, Dalton Trans., 2017, 46, 14002 RSC.
- H. C. Hoffmann, B. Assfour, F. Epperlein, N. Klein, S. Paasch, I. Senkovska, S. Kaskel, G. Seifert and E. Brunner, J. Am. Chem. Soc., 2011, 133, 8681 CrossRef CAS.
- P. S. Petkov, V. Bon, C. L. Hobday, A. B. Kuc, P. Melix, S. Kaskel, T. Düren and T. Heine, Phys. Chem. Chem. Phys., 2019, 21, 674 RSC.
- M. Sin, N. Kavoosi, M. Rauche, J. Pallmann, S. Paasch, I. Senkovska, S. Kaskel and E. Brunner, Langmuir, 2019, 35, 3162 CrossRef CAS.
- M. Mendt, S. Ehrling, I. Senkovska, S. Kaskel and A. Pöppl, Inorg. Chem., 2019, 58, 4561 CrossRef CAS.
- W. Lu, Z. Wei, Z.-Y. Gu, T.-F. Liu, J. Park, J. Park, J. Tian, M. Zhang, Q. Zhang and T. Gentle III, Chem. Soc. Rev., 2014, 43, 5561 RSC.
- E. Brunner, D. Freude, B. Gerstein and H. Pfeifer, J. Magn. Reson., 1990, 90, 90 CAS.
- H. Liu, H.-M. Kao and C. P. Grey, J. Phys. Chem. B, 1999, 103, 4786 CrossRef CAS.
- M. M. Maricq and J. Waugh, J. Chem. Phys., 1979, 70, 3300 CrossRef CAS.
- A. Nayeem and J. P. Yesinowski, J. Chem. Phys., 1988, 89, 4600 CrossRef CAS.
- K. Trepte, S. Schwalbe and G. Seifert, Phys. Chem. Chem. Phys., 2015, 17, 17122 RSC.
- E. Brunner and U. Sternberg, Prog. Nucl. Magn. Reson. Spectrosc., 1998, 32, 21 CrossRef CAS.
- J. Hwang, R. Yan, M. Oschatz and B. V. Schmidt, J. Mater. Chem. A, 2018, 6, 23521 RSC.
- L. Brammer, D. Zhao, F. T. Ladipo and J. Braddock-Wilking, Acta Crystallogr., Sect. B: Struct. Sci., 1995, 51, 632 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details, powder X-ray diffraction patterns, SEM images, details of field- and temperature-dependent NMR experiments and investigation of the solvent effects. See DOI: 10.1039/c9cc04298a |
| ‡ Both authors contributed equally. |
| § Current affilation: Stratingh Institute for Chemistry, University of Groningen, NL-9747 AG Groningen, The Netherlands. |
| This journal is © The Royal Society of Chemistry 2019 |