
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Diborane heterolysis and P(V) reduction by Ph3P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/h2_char_e001.gif) O coordination to magnesium†
O coordination to magnesium†
Anne-Frédérique
Pécharman
,
Nasir A.
Rajabi
,
Michael S.
Hill
 *,
Claire L.
McMullin
* and
Mary F.
Mahon
*,
Claire L.
McMullin
* and
Mary F.
Mahon
Department of Chemistry, University of Bath, Claverton Down, Bath, BA2 7AY, UK. E-mail: msh27@bath.ac.uk; cm2025@bath.ac.uk
First published on 4th July 2019
Abstract
Reaction of a magnesium diboranate complex with triphenylphosphine oxide provides a terminal magnesium boryl, which is itself a potent reagent for the deoxygenative reduction of Ph3PO. Computational analysis with density functional theory (DFT) indicates that B–B bond activation results from initial coordination of the P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond of the phosphine oxide to magnesium.
O bond of the phosphine oxide to magnesium.
The chemistry of nucleophilic group 13-centred anions has been an area of interest for over 20 years. The viability of singlet lithium boryl anions (X2B–Li; X = F, Cl, O) was posited by Schleyer in 1995,1 while the electronic structures of a range of model boron, aluminium, gallium and indium imidazolate anions (1–4) were evaluated by Schoeller and co-workers a short time later.2 Schmidbaur's realisation of the N-heterocyclic gallyl anion (5) in 1999, therefore, was a true landmark in group 13 element synthesis.3 Although the chemistry of compound 5 was never exploited, Jones’ subsequent exploration of the reactivity of the closely-related gallyl (6),4 definitively established the potential of such species as gallium-centred nucleophiles and reducing agents.5–17
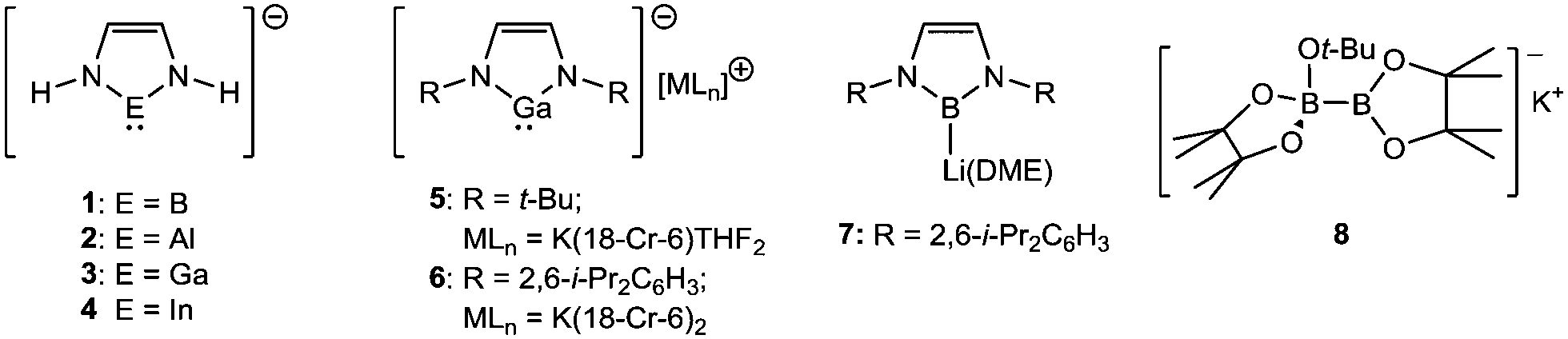
While related aluminium- and indium-centred anions are of a very recent origin,18–21 the veracity of Schleyer's initial speculation was realised in Yamashita and Nozaki's synthesis of the lithium boryl (7) in 2006.22–25 In a similar manner to Jones’ gallium anions, the stability of the carbenoid B(I) centre is based on the combination of kinetic, inductive and mesomeric stability provided by the steric demands of the bulky 2,6-di-iso-propylphenyl (Dipp) and electronegative N-donor substituents, respectively. While the nucleophilic reactivity of compound 7 has been validated toward an array of organic and inorganic electrophiles,26–33 in common with many other subsequently reported boron nucleophiles,34–37 its synthesis requires the synthetically inconvenient reduction of a trivalent boron precursor with a strong alkali metal reducing agent.
During a similar timeframe, the study of desymmetrised diborane derivatives, for example the bis(pinacolato)diborane (B2pin2) derivative 8,38,39 has attracted significant attention.40–44
Applied under either ‘metal-free’ or ‘metal-catalysed’ conditions, such species have been found to provide nucleophilic boron surrogates, albeit without the explicit generation of any observable boryl anion.
Our own research has focused on β-diketiminato (BDI) magnesium compounds such as [(BDI)Mg{pinBB(n-Bu)pin}] (9, Scheme 1).45,46 The [pinBB(n-Bu)pin]− diboranate anion is analogous to that of species 8 and we have observed that compound 9 reacts similarly through elimination of n-BuBpin and as a reagent for nucleophilic B–C and B–B bond formation when treated with C- or B-centred electrophiles.47–49 Although this is consistent with the generation of an intermediate boryl species, we have also observed that reaction of 9 with imines provides the N–B bonded organomagnesium compounds rather than the product of nucleophilic C-borylation (Scheme 1).50 In this case, B–B cleavage does not result in a terminal magnesium boryl species but occurs through attack of the imine nitrogen at the three-coordinate boron of the diboranate anion.50 In contrast, treatment of 9 with 4-dimethylaminopyridine does provide the Mg–B bonded species, [(BDI)Mg(Bpin)DMAP] (10),45,46 raising questions about the generality of such an ‘outer sphere’ process. In this contribution, we extend our study to the reaction of compound 9 with triphenylphosphine oxide and address the mechanism of the resultant B–B heterolysis.
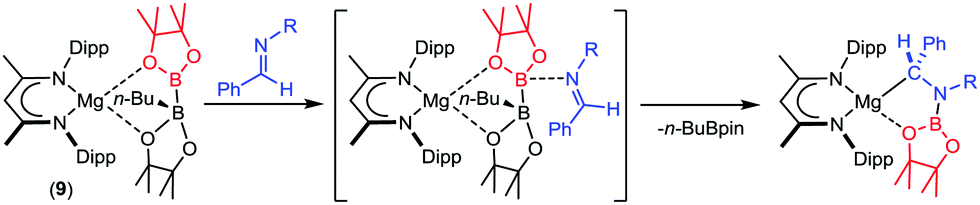 | ||
| Scheme 1 Reaction of compound 9 with aldimines (R – alkyl or aryl). | ||
An equimolar equivalent of Ph3PO was added at room temperature to a d8-toluene solution of compound 9. Immediate analysis by NMR spectroscopy demonstrated the generation of a single new β-diketiminate derivative (11), which was most clearly characterised by the emergence of a BDI methine resonance at δ 4.91 ppm and a signal at δ 35.2 ppm in the respective 1H and 31P{1H} NMR spectra. Although resonances associated with the displacement of neutral n-BuBpin were also observed in both the 1H and 11B NMR spectra, it was clear from continued monitoring that compound 11 itself was subject to a further onward transformation. This was most readily apparent in the 31P{1H} NMR spectrum where a second new species (12) was manifested from the development of a resonance at δ 36.4 ppm. The emergence of compound 12 was accompanied by the simultaneous appearance of a further 31P{1H} resonance with an identical intensity at δ −5.3 ppm, which was readily assigned to triphenylphosphine.51 Although monitoring of these changes to the 31P{1H} NMR spectra over 24 hours confirmed the selective conversion of 11 to compound 12 and Ph3P, inspection of the corresponding 1H NMR data also demonstrated that this process was accompanied by the re-formation of half of the initial compound 9.
The origin of these observations (Scheme 2), was resolved by single crystal X-ray diffraction analysis of both compounds, 11 and 12. Although samples of 12 were readily obtained by fractional crystallisation from a saturated toluene solution of the ultimate reaction mixture, the onward reactivity of 11 made the isolation of an analytically pure sample more problematic. Nevertheless, termination of a further reaction after only one hour provided colourless single crystals, which formed during removal of the reaction solvent under reduced pressure. The resultant solid state structures are illustrated in Fig. 1.
 | ||
| Scheme 2 Synthesis of compounds 11 and 12 from the stoichiometric reaction of compound 9 and Ph3PO and the reductive deoxygenation of Ph3PO. | ||
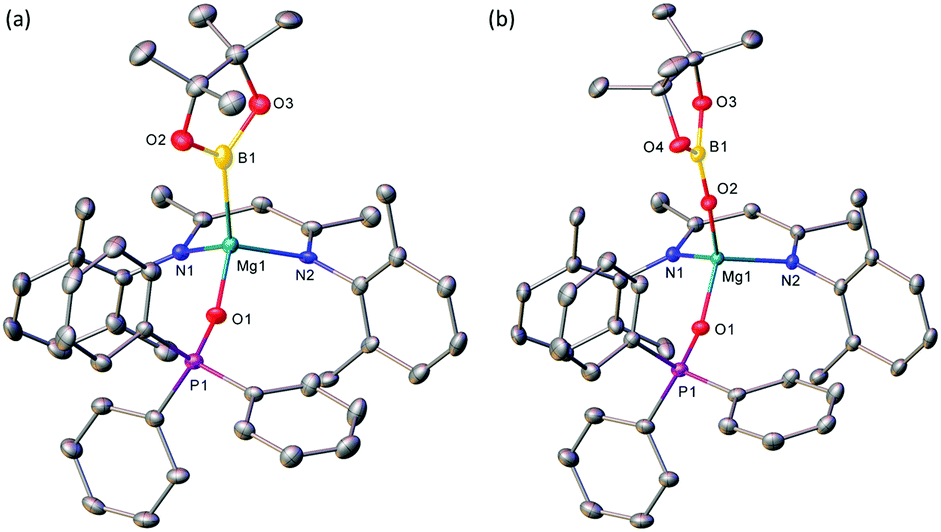 | ||
| Fig. 1 ORTEP representations (30% probability ellipsoids) of the Mg1-containing molecules of (a) compound 11 and (b) compound 12. Hydrogen atoms and iso-propyl methyl groups are removed for clarity. Selected bond lengths (Å) and angles (°): (11) B1–Mg1 2.327(7), B1–O2 1.430(8), B1–O3 1.458(8), Mg1–N1 2.076(4), Mg1–N2 2.076(4), Mg1–O1 1.954(4), P1–O1 1.490(3), N1–Mg1–B1 117.1(2), N1–Mg1–N2 91.48(17), N2–Mg1–B1 115.6(2), O1–Mg1–B1 114.8(2), O1–Mg1–N1 107.71(18), O1–Mg1–N2 107.64(17); (12) B1–O2 1.302(3), B1–O3 1.400(3), B1–O4 1.405(3), Mg1–N1 2.0529(19), Mg1–N2 2.0538(19), Mg1–O1 1.9256(17), Mg1–O2 1.8697(17), O1–P1 1.4989(16), O2–B1–O3 125.4(2), O2–B1–O4 125.2(2), O3–B1–O4 109.4(2), N1–Mg1–N2 92.81(8), O1–Mg1–N1 108.41(8), O1–Mg1–N2 111.31(8), O2–Mg1–N1 119.93(8), O2–Mg1–N2 117.11(8), O2–Mg1–O1 106.69(8). | ||
Compound 11 was identified as a four-coordinate terminal magnesium boryl derivative. A molecule of Ph3PO fulfils a similar role to the unidentate DMAP ligand in 10,45,46 while the three remaining magnesium-to-ligand contacts are similarly provided by the nitrogen atoms of the BDI ligand and a single terminal Bpin unit. The Mg1–B1 distance [2.327(7) Å] observed in compound 11 is effectively identical to that determined for [(BDI)Mg(Bpin)DMAP] (10) [2.324(2) Å] and the further DMAP-coordinated species, [(BDI)Mg(Bhex)DMAP] [2.319(3) Å], which was synthesised by analogous Mg-centred heterolysis of 4,4,4′,4′,6,6′-hexamethyl-2,2′-bi(1,3,2-dioxaborinane) (B2hex2).46 These measurements also lie within the range seen for three reported magnesium derivatives synthesised by reactions of compound 7 with MgBr2 [2.281(6)–2.377(4) Å].26
While three of the coordination sites of compound 12 are again occupied by the bidentate BDI ligand and a molecule of Ph3PO, the fourth is provided by a terminal pinacolatoboryloxide unit. The structure of compound 12 [Mg1–O1 1.9256(17), Mg2–O5 1.9188(17) Å] is, thus, reminiscent of two previously described magnesium diphenylboryloxide complexes, [(BDI)Mg(OBPh2)(n-BuBpin)] [Mg–O 1.8492(16) Å] and [(BDI)Mg(OBPh2)DMAP] [Mg–O 1.856(3) Å].48 These latter compounds were synthesised by the respective reactions of compounds 9 and 10 with diphenylborinic anhydride as the source of the {OBPh2} ligand. In contrast, the boryloxide ligand of 12 arises from formal insertion of an oxygen atom into the Mg–B bond of compound 11. Although compound 11 clearly holds the potential for direct oxygenation through the ingress of atmospheric oxygen, this possibility may be discounted by the overall stoichiometry and notable selectivity of the reaction (Scheme 2).
Although the selective deoxygenation of small molecule oxygenates including CO2 by copper and rhodium pinacolatoboryl species has been described,52–54 and the catalytic and stoichiometric reduction of secondary and tertiary phosphine oxides to phosphines has significant precedent,55 the current transformation appears to be unique. The reaction stoichiometry displayed in Scheme 2 implies the establishment of an equilibrium between Ph3PO and the displaced molecule of n-BuBpin such that the generation of every molecule of compound 12 requires two molecules of phosphine oxide. On this basis, a further reaction was performed in a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 stoichiometry between Ph3PO and compound 9. Although the initial formation of the boryl derivative (11) was again clearly evident by 31P{1H} NMR spectroscopy (δ 35.2 ppm), this signal was observed with a resonance of equal intensity associated with the unreacted equivalent of Ph3PO (δ 24.1 ppm). Continued monitoring of this reaction over 24 hours again evidenced the production of Ph3P, which occurred simultaneously with the complete transformation of 9 to compound 12. The dynamic nature of the magnesium-to-Ph3PO and magnesium-to-DMAP interactions was also underscored by a further reaction performed between equimolar quantities of [(BDI)Mg(Bpin)DMAP] (10) and Ph3PO, which provided a mixture of compounds 10–12 alongside free DMAP and Ph3PO (Fig. S9–S11, ESI†).
:1 stoichiometry between Ph3PO and compound 9. Although the initial formation of the boryl derivative (11) was again clearly evident by 31P{1H} NMR spectroscopy (δ 35.2 ppm), this signal was observed with a resonance of equal intensity associated with the unreacted equivalent of Ph3PO (δ 24.1 ppm). Continued monitoring of this reaction over 24 hours again evidenced the production of Ph3P, which occurred simultaneously with the complete transformation of 9 to compound 12. The dynamic nature of the magnesium-to-Ph3PO and magnesium-to-DMAP interactions was also underscored by a further reaction performed between equimolar quantities of [(BDI)Mg(Bpin)DMAP] (10) and Ph3PO, which provided a mixture of compounds 10–12 alongside free DMAP and Ph3PO (Fig. S9–S11, ESI†).
The origins of the B–B and PO bond breaking processes were investigated by DFT (BP86 optimised, see ESI,† for full methodology details). Initial calculations assessed the potential for spontaneous cleavage of the B–B bond of compound 9 in the absence of any external reagent. Although the generation of a putative three-coordinate boryl species, [(BDI)MgBpin] (shown as species E in Fig. S12, ESI†) was found to be endergonic by 12.9 kcal mol−1, the facility of n-BuBpin elimination was emphasised by the location of a transition state, [TS(9-D)], describing a barrier toward B–B bond breaking of only 19.4 kcal mol−1. Incorporation of Ph3PO into the reaction profile led us to address three scenarios invoking the attack of the phosphine oxide either at the Mg centre or the three-coordinate boron of the [pinBB(n-Bu)pin]− anion. While the latter of these possibilities yielded a credible transition state, [TS(G-H)], the barrier associated with this ‘outer sphere’ process was 23.2 kcal mol−1 and led to the direct reduction and deoxygenation of the phosphine oxide reagent circumventing the production of compound 11 (Fig. S13, ESI†). Addition of Ph3PO to species E, however, indicated that the formation of compound 11 is mildly exergonic (ΔG = −3.6 kcal mol−1, Fig. S12, ESI†). Although this process enables the construction of a plausible profile for the subsequent conversion of compound 11 to 12 (vide infra), further assessment indicated the existence of the more kinetically-accessible pathway shown in Fig. 2.
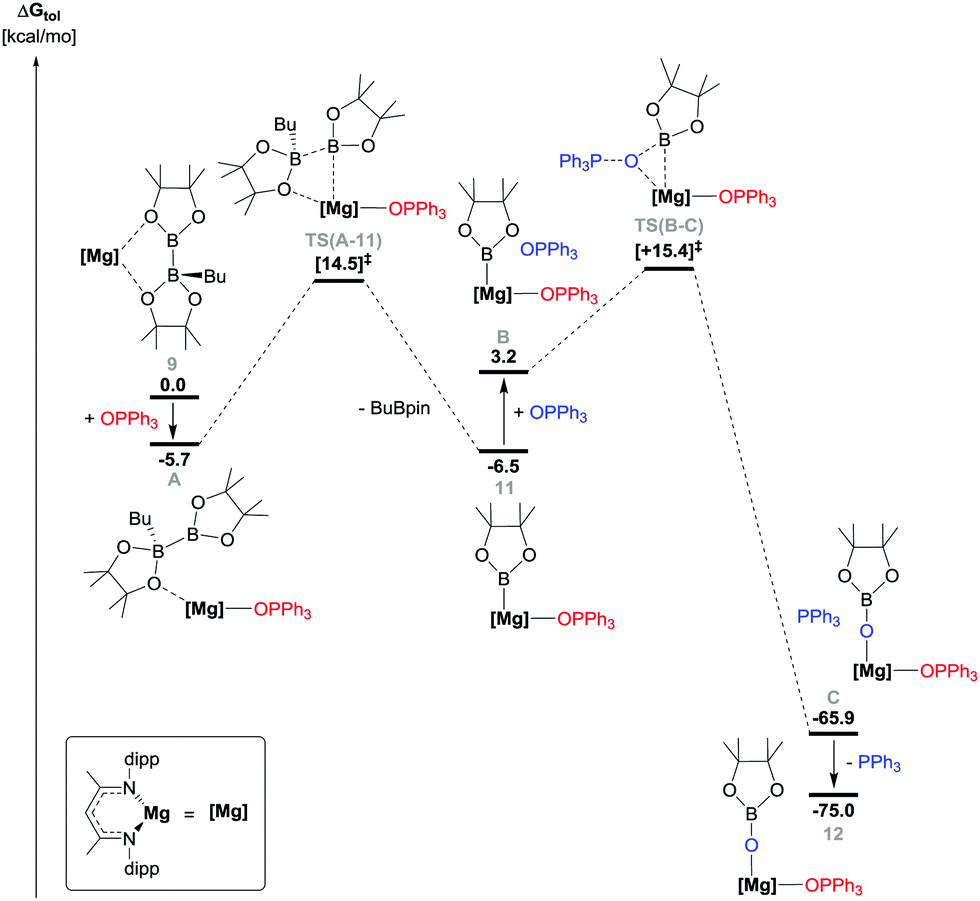 | ||
| Fig. 2 DFT (BP86-D3BJ(BS2,toluene)//BP86(BS1)) calculated free energy (kcal mol−1) profile for the reaction of Ph3PO with compound 9 (in toluene), relative to 9 and the free substrates. | ||
Coordination of Ph3PO to the Mg centre of compound 9 necessitates the formation of an adduct species (A) and the decoordination of the trigonal pinB component of the [pinBB(n-Bu)pin]− anion. Although entropically-disfavoured, this process is exergonic (ΔG = −5.7 kcal mol−1) and is facilitated by the low steric demands of the PO donor. Consistent with the spontaneous generation of 11 at room temperature, this process induces the rupture of the B–B bond via transition state TS(A-11), which is located 14.5 kcal mol−1 above compound 9.
The subsequent transformation of 11 to compound 12 is significantly exergonic (ΔG = −68.5 kcal mol−1) and involves the traversal of a barrier, [TS(B-C)], of only 12.2 kcal mol−1. Formal P(V) reduction is facilitated via the assembly of a three-centre Mg–O–B interaction at this transition state en route to species C, which ensues with simultaneous P–O bond cleavage and the generation of the requisite Mg–O and O–B bonds.56
In conclusion, treatment of compound 9 with Ph3PO provides a magnesium boryl derivative by inner sphere coordination of the PO bond to magnesium. This deduction, therefore, contrasts with the outer sphere attack at boron implicated in reactions of 9 with more sterically encumbered imine reagents (Scheme 1).50 In both cases, however, B–B bond cleavage is facile, indicating that the mode of [pinBB(n-Bu)pin]− heterolysis is subtly dependent on the degree of kinetic discrimination provided by the co-reagent. We are, thus, continuing to examine the implications of these observations for a wider range of substrates and element centres.
We thank the EPSRC (UK) for their generous support of this research (Grants No. EP/N014456/1 and EP/R020752/1). This research made use of the Balena High Performance Computing (HPC) Service at the University of Bath. The referees are thanked for their insightful comments with regard to the computed reaction profile shown in Fig. 2.
Conflicts of interest
There are no conflicts to declare.Notes and references
- M. Wagner, N. Hommes, H. Noth and P. V. Schleyer, Inorg. Chem., 1995, 34, 607–614 CrossRef CAS
.
- A. Sundermann, M. Reiher and W. W. Schoeller, Eur. J. Inorg. Chem., 1998, 305–310 CrossRef CAS
- E. S. Schmidt, A. Jockisch and H. Schmidbaur, J. Am. Chem. Soc., 1999, 121, 9758–9759 CrossRef CAS
- R. J. Baker, R. D. Farley, C. Jones, M. Kloth and D. M. Murphy, J. Chem. Soc., Dalton Trans., 2002, 3844–3850 RSC
- R. J. Baker, C. Jones, M. Kloth and J. A. Platts, Angew. Chem., Int. Ed., 2003, 42, 2660–2663 CrossRef CAS PubMed
- R. J. Baker, C. Jones and J. A. Platts, J. Am. Chem. Soc., 2003, 125, 10534–10535 CrossRef CAS
- R. J. Baker, C. Jones, M. Kloth and J. A. Platts, Organometallics, 2004, 23, 4811–4813 CrossRef CAS
- R. J. Baker, C. Jones and M. Kloth, Dalton Trans., 2005, 2106–2110 RSC
- S. Aldridge, R. J. Baker, N. D. Coombs, C. Jones, R. P. Rose, A. Rossin and D. J. Willock, Dalton Trans., 2006, 3313–3320 RSC
- C. Jones, D. P. Mills, J. A. Platts and R. P. Rose, Inorg. Chem., 2006, 45, 3146–3148 CrossRef CAS
- C. Jones, R. P. Rose and A. Stasch, Dalton Trans., 2007, 2997–2999 RSC
- S. P. Green, C. Jones, K. A. Lippert, D. P. Mills and A. Stasch, Inorg. Chem., 2006, 45, 7242–7251 CrossRef CAS
- P. L. Arnold, S. T. Liddle, J. McMaster, C. Jones and D. P. Mills, J. Am. Chem. Soc., 2007, 129, 5360–5361 CrossRef CAS
- C. Jones, D. P. Mills, R. P. Rose and A. Stasch, Dalton Trans., 2008, 4395–4408 RSC
- C. Jones, D. P. Mills, R. P. Rose, A. Stasch and W. D. Woodul, J. Organomet. Chem., 2010, 695, 2410–2417 CrossRef CAS
- S. T. Liddle, J. McMaster, D. P. Mills, A. J. Blake, C. Jones and W. D. Woodul, Angew. Chem., Int. Ed., 2009, 48, 1077–1080 CrossRef CAS
- O. Bonello, C. Jones, A. Stasch and W. D. Woodul, Organometallics, 2010, 29, 4914–4922 CrossRef CAS
- J. Hicks, P. Vasko, J. M. Goicoechea and S. Aldridge, Nature, 2018, 557, 92–95 CrossRef CAS
- J. Hicks, A. Mansikkamaki, P. Vasko, J. M. Goicoechea and S. Aldridge, Nat. Chem., 2019, 11, 237–241 CrossRef CAS
- R. J. Schwamm, M. D. Anker, M. Lein, M. P. Coles and C. M. Fitchett, Angew. Chem., Int. Ed., 2018, 57, 5885–5887 CrossRef CAS
- R. J. Schwamm, M. D. Anker, M. Lein and M. P. Coles, Angew. Chem., Int. Ed., 2019, 58, 1489–1493 CrossRef CAS
- Y. Segawa, M. Yamashita and K. Nozaki, Science, 2006, 314, 113–115 CrossRef CAS
- Y. Segawa, Y. Suzuki, M. Yamashita and K. Nozaki, J. Am. Chem. Soc., 2008, 130, 16069–16079 CrossRef CAS
- M. Yamashita and K. Nozaki, J. Synth. Org. Chem., Jap., 2010, 68, 359–369 CrossRef CAS
-
M. Yamashita and K. Nozaki, in Synthesis and Application of Organoboron Compounds, ed. E. Fernandez and A. Whiting, 2015, vol. 49, pp. 1–37 Search PubMed
- M. Yamashita, Y. Suzuki, Y. Segawa and K. Nozaki, J. Am. Chem. Soc., 2007, 129, 9570–9571 CrossRef CAS
- Y. Segawa, M. Yamashita and K. Nozaki, Angew. Chem., Int. Ed., 2007, 46, 6710–6713 CrossRef CAS
- T. Terabayashi, T. Kajiwara, M. Yamashita and K. Nozaki, J. Am. Chem. Soc., 2009, 131, 14162–14163 CrossRef CAS
- T. Arnold, H. Braunschweig, W. C. Ewing, T. Kramer, J. Mies and J. K. Schuster, Chem. Commun., 2015, 51, 737–740 RSC
- A. V. Protchenko, K. H. Birjkumar, D. Dange, A. D. Schwarz, D. Vidovic, C. Jones, N. Kaltsoyannis, P. Mountford and S. Aldridge, J. Am. Chem. Soc., 2012, 134, 6500–6503 CrossRef CAS
- A. V. Protchenko, D. Dange, M. P. Blake, A. D. Schwarz, C. Jones, P. Mountford and S. Aldridge, J. Am. Chem. Soc., 2014, 136, 10902–10905 CrossRef CAS PubMed
- A. V. Protchenko, D. Dange, J. R. Harmer, C. Y. Tang, A. D. Schwarz, M. J. Kelly, N. Phillips, R. Tirfoin, K. H. Birjkumar, C. Jones, N. Kaltsoyannis, P. Mountford and S. Aldridge, Nat. Chem., 2014, 6, 315–319 CrossRef CAS
- D. Dange, A. Davey, J. A. B. Abdalla, S. Aldridge and C. Jones, Chem. Commun., 2015, 51, 7128–7131 RSC
- B. L. Wang, Y. X. Li, R. Ganguly, H. Hirao and R. Kinjo, Nat. Commun., 2016, 7, 11871 CrossRef CAS
- W. Lu, H. T. Hu, Y. X. Li, R. Ganguly and R. Kinjo, J. Am. Chem. Soc., 2016, 138, 6650–6661 CrossRef CAS
- H. Braunschweig, C. W. Chiu, K. Radacki and T. Kupfer, Angew. Chem., Int. Ed., 2010, 49, 2041–2044 CrossRef CAS
- R. Kinjo, B. Donnadieu, M. A. Celik, G. Frenking and G. Bertrand, Science, 2011, 333, 610–613 CrossRef CAS
- S. Pietsch, E. C. Neeve, D. C. Apperley, R. Bertermann, F. Y. Mo, D. Qiu, M. S. Cheung, L. Dang, J. B. Wang, U. Radius, Z. Y. Lin, C. Kleeberg and T. B. Marder, Chem. – Eur. J., 2015, 21, 7082–7098 CrossRef CAS
- M. Eck, S. Wrtemberger-Pietsch, A. Eichhorn, J. H. J. Berthel, R. Bertermann, U. S. D. Paul, H. Schneider, A. Friedrich, C. Kleeberg, U. Radius and T. B. Marder, Dalton Trans., 2017, 46, 3661–3680 RSC
- H. Gulyas, A. Bonet, C. Pubill-Ulldemolins, C. Sole, J. Cid and E. Fernandez, Pure Appl. Chem., 2012, 84, 2219–2231 CAS
- J. Cid, H. Gulyas, J. J. Carbo and E. Fernandez, Chem. Soc. Rev., 2012, 41, 3558–3570 RSC
- R. D. Dewhurst, E. C. Neeve, H. Braunschweig and T. B. Marder, Chem. Commun., 2015, 51, 9594–9607 RSC
- E. C. Neeve, S. J. Geier, I. A. I. Mkhalid, S. A. Westcott and T. B. Marder, Chem. Rev., 2016, 116, 9091–9161 CrossRef CAS
- T. Kaese, T. Trageser, H. Budy, M. Bolte, H.-W. Lerner and M. Wagner, Chem. Sci., 2018, 9, 3881–3891 RSC
- A. F. Pécharman, A. L. Colebatch, M. S. Hill, C. L. McMullin, M. F. Mahon and C. Weetman, Nat. Commun., 2017, 8, 15022 CrossRef
- A. F. Pécharman, M. S. Hill and M. F. Mahon, Dalton Trans., 2018, 47, 7300–7305 RSC
- A. F. Pécharman, M. S. Hill, C. L. McMullin and M. F. Mahon, Angew. Chem., Int. Ed., 2017, 56, 16363–16366 CrossRef
- A. F. Pécharman, M. S. Hill and M. F. Mahon, Angew. Chem., Int. Ed., 2018, 57, 10688–10691 CrossRef
- A.-F. Pécharman, M. S. Hill, C. L. McMullin and M. F. Mahon, Organometallics, 2018, 37, 4457–4464 CrossRef
- A. F. Pécharman, M. S. Hill, G. McMullon, C. L. McMullin and M. F. Mahon, Chem. Sci., 2019 10.1039/C9SC02087J
- A. D. Hunter, T. R. Williams, B. M. Zarzyczny, H. W. Bottesch, S. A. Dolan, K. A. McDowell, D. N. Thomas and C. H. Mahler, Organometallics, 2016, 35, 2701–2706 CrossRef CAS
- D. S. Laitar, P. Muller and J. P. Sadighi, J. Am. Chem. Soc., 2005, 127, 17196–17197 CrossRef CAS
- S. I. Kallane, T. Braun, B. Braun and S. Mebs, Dalton Trans., 2014, 43, 6786–6801 RSC
- S. I. Kallane, T. Braun, M. Teltewskoi, B. Braun, R. Herrmann and R. Laubenstein, Chem. Commun., 2015, 51, 14613–14616 RSC
- D. Herault, D. H. Nguyen, D. Nuel and G. Buono, Chem. Soc. Rev., 2015, 44, 2508–2528 RSC
- The reaction stoichiometry shown in Scheme 2 is, thus, explicable as the barrier associated with the reformation of 9 from 11 (21.0 kcal mol−1) is competitive with the onward transformation of 11 to compound 12 (21.9 kcal mol−1).
Footnote |
| † Electronic supplementary information (ESI) available: General synthetic experimental details, NMR spectra, details of the X-ray analysis of compounds 11 and 12, details of the computational analysis and atomic coordinates of the DFT computed structures. CCDC 1915140 (11) and 1915141 (12). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9cc04294f |
| This journal is © The Royal Society of Chemistry 2019 |