
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBeacon-mediated exponential amplification reaction (BEAR) using a single enzyme and primer†
Ashley M.
Newbigging
 ,
Hongquan
Zhang
and
X. Chris
Le
*
,
Hongquan
Zhang
and
X. Chris
Le
*
Department of Laboratory Medicine and Pathology, University of Alberta, Edmonton, Alberta, Canada. E-mail: xcle@ualberta.ca
First published on 19th August 2019
Abstract
Beacon-mediated Exponential Amplification Reaction (BEAR) enables isothermal, exponential signal amplification. BEAR uses only a single enzyme and a single primer. Detection of 0.2 amol of a mitochondrial DNA with a point mutation in less than an hour demonstrates an application of the BEAR technique for nucleic acid research.
Polymerase chain reaction (PCR) is powerful for the amplification of nucleic acids. Each cycle of PCR requires changes in temperatures between lower temperatures for annealing (e.g., 60 °C) and extension (e.g., 70 °C), and a higher temperature for denaturation (e.g., 95 °C) of double stranded DNA (dsDNA) to single stranded DNA (ssDNA). Eliminating thermal cycling would allow the use of simpler devices, and facilitate the potential for point-of-care testing particularly in resource limited settings. Several isothermal and exponential amplification techniques have been developed,1 such as exponential strand displacement amplification (E-SDA),2 exponential rolling circle amplification (E-RCA),3 exponential amplification reaction (EXPAR),4 nucleic acid sequence based amplification (NASBA),5 helicase-dependent amplification (HDA),6 and recombinase polymerase amplification (RPA).7 Although there is no need for these techniques to use a high temperature for denaturation, they require enzymes in addition to the polymerase, to help generate ssDNA from dsDNA. EXPAR and SDA8 use the cooperation of an endonuclease and a polymerase to generate ssDNA. HDA and RPA use helicase and recombinase, respectively, to convert dsDNA to ssDNA. One exception is loop-mediated isothermal amplification (LAMP),9 which uses only a polymerase. However, LAMP requires four to six primers, which complicates the design.
Limiting the number of required enzymes and primers would simplify technical procedures and reduce the restrictions on reagent storage requirements, both of which can extend the applicability of amplification techniques for use in point-of-care testing. Despite many recent advances in isothermal methods with exponential amplification,10 there is no technique that uses a single enzyme and a single primer for isothermal and exponential amplification.
We introduce a new technique, Beacon-mediated Exponential Amplification Reaction (BEAR), for the detection of nucleic acid targets using isothermal and exponential signal amplification with a single enzyme and a single primer (Scheme 1). Key to the BEAR technique is a specially designed DNA beacon, the amplifiable beacon, that cages a primer-binding region. The target nucleic acid interacts with the amplifiable beacon and opens the stem, uncaging this region for the primer to bind and initiate polymerase extension. The target strand and an additional fluorophore-conjugated strand (FS) are displaced from the beacon and the displaced FS produces a fluorescence signal. The target is recycled and interacts with other amplifiable beacons to initiate additional reactions, which would result in linear amplification. To enable exponential amplification of the detection signal, we designed the displaced FS to also interact with amplifiable beacons to initiate the amplification reaction. Thus, after being displaced, the FS serves as a reporter and also initiates more reactions.
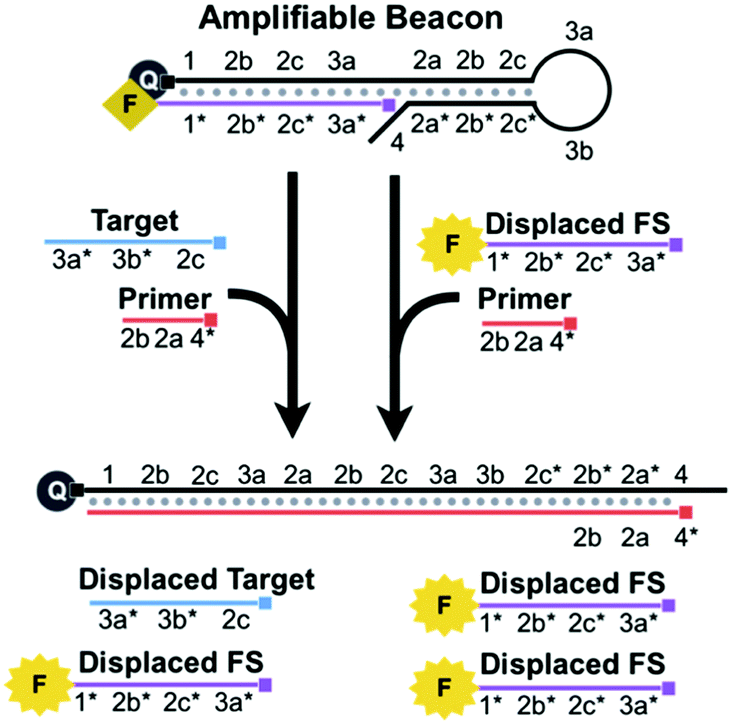 | ||
| Scheme 1 BEAR uses an amplifiable beacon, a single enzyme, and a single primer to produce amplified fluorescence signals in response to a nucleic acid target. The target and an additional fluorescent strand, FS, are displaced. Exponential amplification is achieved because both the displaced target and displaced FS cycle back to react with the amplifiable beacon. The small square at the end of the DNA sequences indicates the 5′-end. | ||
For the ease of explanation of our design, complementary domains are denoted with an asterisk (e.g. domain 1* is complementary to domain 1) (Scheme S1, ESI†). The amplifiable beacon is created by annealing the FS to the long overhang of a DNA hairpin, HP (Scheme S2, ESI†). A fluorescence quencher is conjugated to the 5′ end of HP and a fluorophore is conjugated to the 3′ end of the FS. When HP and FS are annealed together to form the beacon, the fluorescence is quenched. The sequence of HP contains functional loop (3a and 3b), stem (2a/2a*, 2b/2b*, and 2c/2c*), primer-binding (4, 2a*, and 2b*), and long 5′ overhang (1, 2b, 2c, and 3a) domains. We designed the 14-bp stem to cage a portion of the primer-binding region, 2a and 2b*, and thus this region is unavailable for the primer to bind.
While 4 is complementary to part of the primer, the main intention of designing this short overhang is to prevent extension via the polymerase which would result in target-independent displacement of the FS.
BEAR begins when a nucleic acid target binds to 3a, 3b, and 2c* of the loop and stem of HP within an amplifiable beacon (Scheme 2A). The stem opens and exposes 2a* and 2b* to the primer. The subsequent binding of the primer to 4, 2a*, and 2b* initiates 5′ to 3′ extension via a DNA polymerase, using HP as the template. The polymerase has strong strand displacement activity and displaces the target and the FS that comprised the amplifiable beacon. When the FS is displaced from HP, the fluorophore and quencher are separated and the fluorescence is restored. After complete polymerase extension, HP has no further purpose in the reaction and is waste. The displaced target is cycled back to initiate new reactions by binding to other amplifiable beacons.
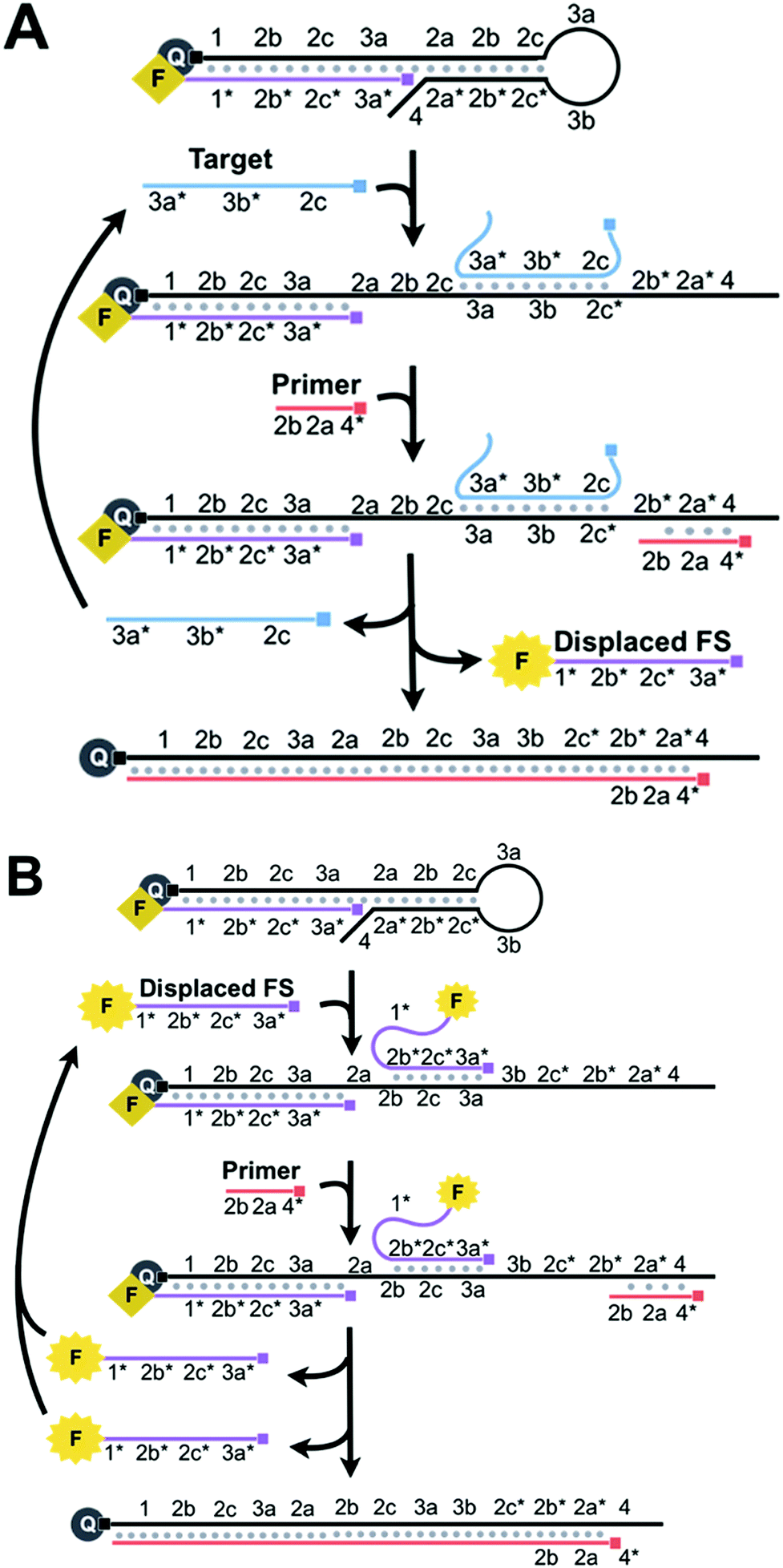 | ||
| Scheme 2 (A) Binding of the target to the loop and stem of HP (3a, 3b, and 2c*) opens the stem to uncage the primer-binding domains (2a* and 2b*). Primer binding and subsequent extension via the polymerase displace the target and the previously annealed FS. The fluorescence of the FS is restored as it is separated from the quencher. The target is recycled and thus amplification is achieved. (B) The displaced FS also binds to the loop and stem of HP (3a, 2c, and 2b) to open the stem and initiate the primer extension. Primer binding and subsequent extension via the polymerase displace both copies of the FS. The displaced FS is fluorescent and is also recycled for the amplification reactions. Thus, the cyclic reactions involving both the target and the displaced FS result in the exponential amplification. | ||
We achieve exponential amplification of the detection signal by allowing the displaced FS strands also initiate additional reactions (Scheme 2B). This is because we designed the FS to complement both the overhang of HP and the loop and stem of HP. Although the fluorescence of the FS bound to the overhang of HP is quenched, the FS bound to the loop and stem of HP retains its fluorescence. Similar to the reaction initiated by the target, binding of the FS to the loop and stem domains (3a, 2c, and 2b) of HP exposes 2a* and 2b* to the primer. The polymerase extends the primer and displaces both the initiating FS and the previously bound FS, restoring the fluorescence of the FS. Both displaced FS strands can further bind to other beacons to initiate additional reactions. The cyclic reactions initiated by the target and then by the displaced FS result in exponential amplification.
As a proof-of-concept, we applied our technique to detect the point mutation, 8344A > G, in mitochondrial DNA (mtDNA), as a 25-nt target.11 This point mutation is associated with Myoclonus Epilepsy with Ragged Red Fibres (MERRF). The Seitz group has shown that the conformational constraint of the beacon can discriminate mismatches.12
For the formation of the amplifiable beacon, the FS must be bound to the 5′ overhang of HP instead of the loop and stem, as the FS is complementary to both. If the FS binds the loop and stem in the absence of the target, the resulting reaction would produce background. To drive the binding of the FS to the overhang, we increased the complementarity of the FS to the overhang relative to that of the loop and stem by extending the length of 1 on the overhang.
We tested the annealing of the FS to HP using various ratios of [HP] to [FS]. We separated the annealing products from each of these reaction mixtures using polyacrylamide gel electrophoresis (PAGE) (Fig. 1). Product (iii) corresponds to HP, and the increasing band intensity corresponds to increasing amounts of HP relative to the FS. Product (ii) is the correctly formed amplifiable beacon whose fluorescence is completely quenched. When the amount of FS is in excess of HP in a ratio of 1.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, product (i) is detectable, representing the annealed amplifiable beacon with an additional FS bound to the loop and stem. Adding the FS in excess of HP in a ratio of 1.5:1 to force the FS to bind the loop and stem of the beacon supports the fact that domain 1 is sufficient to drive the binding of the FS to the overhang of HP. We further optimized the ratios of [HP] to [FS] to maximize amplification of the target while minimizing the background (Fig. S1, ESI†).
:1, product (i) is detectable, representing the annealed amplifiable beacon with an additional FS bound to the loop and stem. Adding the FS in excess of HP in a ratio of 1.5:1 to force the FS to bind the loop and stem of the beacon supports the fact that domain 1 is sufficient to drive the binding of the FS to the overhang of HP. We further optimized the ratios of [HP] to [FS] to maximize amplification of the target while minimizing the background (Fig. S1, ESI†).
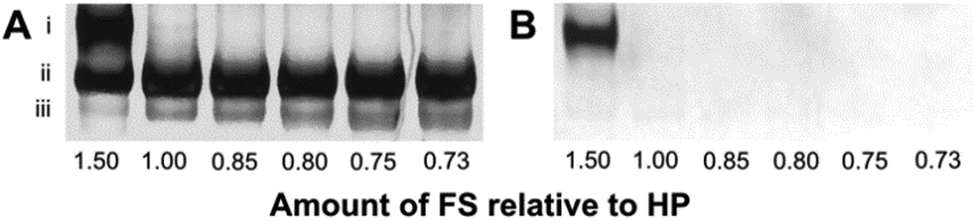 | ||
| Fig. 1 Native PAGE analyses of amplifiable beacon solutions containing various ratios of [HP] to [FS] (1:1.5, 1:1.0, 1:0.85, 1:0.80, 1:0.75, and 1:0.73). The 10% native gel was imaged for FAM fluorescence (B), then stained with SYBR Gold (A) and imaged again. All products are therefore visualized in image A, and only products that have unquenched fluorescence are visualized in image B. The bands correspond to: (i) amplifiable beacon bound to the FS, (ii) amplifiable beacon, and (iii) HP. | ||
Fig. S2, ESI,† shows the typical amplification curves of the BEAR. The sigmoidal shape of the curves generated by 100 pM of the synthetic MERRF target sequence and blank demonstrate characteristic exponential amplification. Similar to other techniques with exponential amplification, three phases are observed: lag, exponential, and plateau. The reaction at 0 min contains primarily amplifiable beacons, characterizing the lag phase. In the lag phase, there is a small number of initiating molecules (target or displaced FS), which results in the slow propagation of the reaction and fluorescence output below the detection limit of the fluorometer. The exponential phase, at ∼26–54 min, generates substantial fluorescence because of the increased amount of displaced FS. Both the lag phase and exponential phase consist of amplifiable beacons that are readily available in abundance for the target or displaced FS to bind to for the propagation of the reaction. The plateau phase is a result of the exhaustion of available amplifiable beacons.
The background signal may arise from the leakage of DNA structures as a result of incomplete chemical synthesis. Leakage may cause improper formation of amplifiable beacons and may result in target-independent displacement of the FS that is then exponentially amplified. We have considered various sources of background (Section 8, ESI†) and minimized background in the optimization processes.
We detected the amplification products at various amplification time points by sampling the reaction mixture and separating the components using PAGE (Fig. S3, ESI†). The amplifiable beacon and reaction products, including the displaced FS, transient complexes, and waste, were detected. The predicted products were detected in each phase: lag (0–28 min), exponential (∼32–46 min), and plateau (after 48 min).
The primer binding location on HP is partially caged within the stem, 2b* and 2a*, and partially exposed, 4. To determine the optimal primer length, we designed two sets of primers (Section 5, ESI†). In one set, we fixed the sequence of the primer on the 5′ end (Primer 5-n) and changed the length of the domain complementary to the stem of HP (2b* and 2a*). In the second set, we fixed the sequence of the primer on the 3′ end (Primer 3-n) and changed the length of 4* (Scheme S3, ESI†). We also tested a full-length primer of 11-nt (Primer 11).
Fig. S4, ESI,† shows that the full-length primer, Primer 11, produced a fast reaction in both the target and the blank, resulting in a small ΔTt (difference between Ttblank and Tttarget). As expected, both primer sets resulted in slower reactions with decreasing primer lengths, which implies that the duplex stability of the bound primer contributes to the rate of the reaction. The shortest primer in each set produced the largest ΔTt. Primers 5–8 and 3–8 showed no substantial difference in ΔTt, indicating that the binding position of the primer does not substantially affect ΔTt. For subsequent experiments, we used Primers 5–8. We also optimized the ratio of HP to FS in forming the amplifiable beacon (Fig S1, ESI†), reaction temperature (Fig. S5, ESI†), polymerase concentrations (Fig. S6, ESI†), complementary domain (Section 9, ESI†), and buffer concentrations.
We assessed the dynamic range of BEAR using various concentrations of the MERRF target sequence, from 1 nM to 10 fM (Fig. 2 and Fig. S7, ESI†). We also obtained the limit of detection (LOD) by testing seven replicate blanks and comparing it to our calibration. The LOD, defined as the concentration equivalent to three times the standard deviation of blanks plus the background signal (3SDblank + Ttblank), was about 10 fM (0.2 amol).
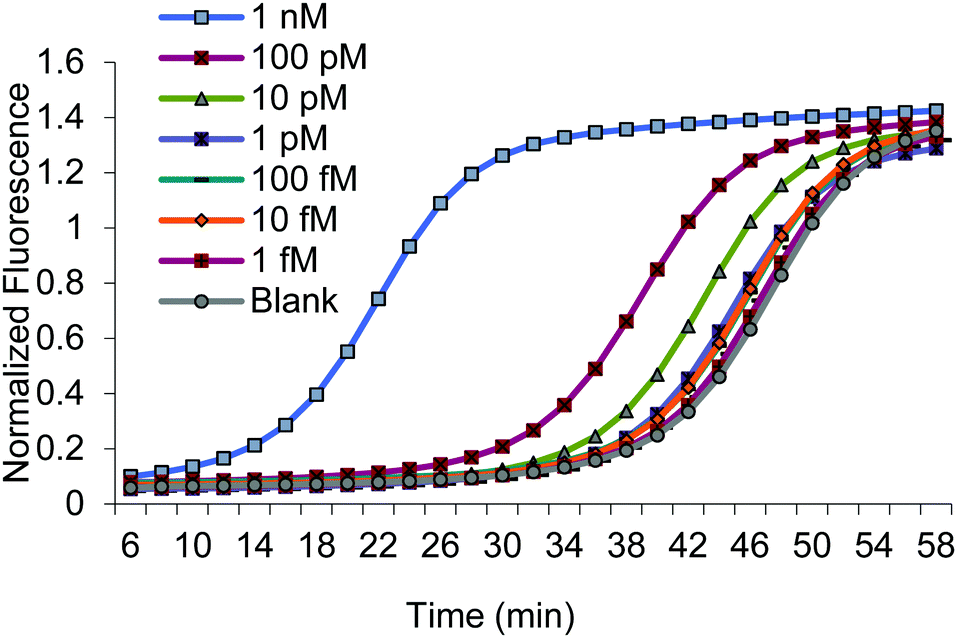 | ||
| Fig. 2 Amplification curves using various concentrations of the MERRF target. | ||
We also investigated the specificity of BEAR by assessing the detection of the MERRF target sequence and the clinical negative control (NC), the normal mtDNA sequence. We compared the reaction of 100 pM of the MERRF target with 100 pM of the clinical negative control (Fig. 3). The BEAR achieved a discrimination factor (DF) of 16.
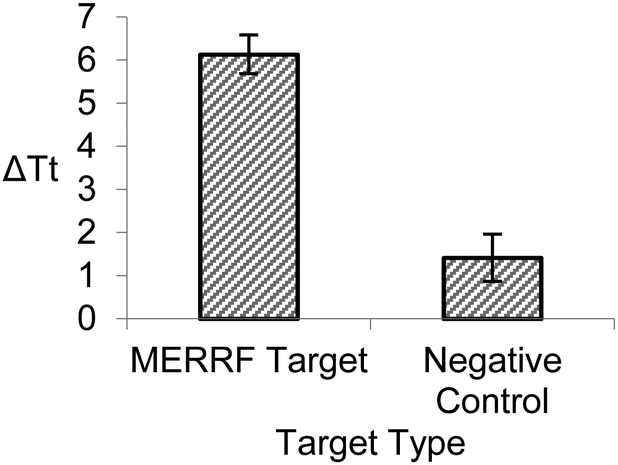 | ||
| Fig. 3 Comparison between 100 pM of the MERRF target and 100 pM of the clinical negative control. ΔTt is the difference in time at threshold between the target and blank. Error bars represent standard deviation from triplicate analyses. | ||
We applied the BEAR technique to the detection of the 10 pM MERRF target in cell lysate (Fig. S9 and Section 10, ESI†). The measured concentration was 9.1 ± 0.8 pM, consistent with the expected concentration and representing an average recovery of 91%.
Our results demonstrate that with a single enzyme and a single primer the BEAR technique is able to achieve isothermal and exponential amplification of the detection signal for a clinically relevant mutation target. Using a single polymerase and a single primer simplifies the technical procedure, which is valuable for point-of-care testing and on-site analysis. BEAR is particularly appropriate for the detection of short nucleic acid targets, as we have demonstrated the detection of a 25-nt MERRF target. Future applications could also include the detection of microRNA (miRNA). The short length of miRNAs poses challenges in accommodating more than one primer as is often required in other techniques.
We thank NSERC, CIHR, Alberta Health, and Alberta Innovates for financial support.
Conflicts of interest
There are no conflicts to declare.References
- L. Yan, J. Zhou, Y. Zheng, A. S. Gamson, B. T. Roembke, S. Nakayama and H. O. Sintim, Mol. BioSyst., 2014, 10, 970 RSC; J. Kim and C. J. Easley, Bioanalysis, 2011, 3, 227 CrossRef CAS PubMed.
- Z. Zhang and C. Zhang, Anal. Chem., 2012, 84, 1623 CrossRef CAS PubMed; C. Shi, Q. Liu, C. Ma and W. Zhong, Anal. Chem., 2014, 86, 336 CrossRef PubMed.
- L. R. Zhang, G. Zhu and C.-Y. Zhang, Anal. Chem., 2014, 86, 6703 CrossRef CAS PubMed; H. Jiang, Y. Xu, L. Dai, X. Liu and D. Kong, Sens. Actuators, B, 2018, 260, 70 CrossRef; W. Zhao, M. M. Ali, M. A. Brook and Y. Li, Angew. Chem., Int. Ed., 2008, 47, 6330 CrossRef PubMed; Q. Xue, Y. Kong, H. Wang and W. Jiang, Chem. Commun., 2017, 53, 10772 RSC; Q. Wang, H. Zheng, X. Gao, Z. Lin and G. Chen, Chem. Commun., 2013, 49, 11418 RSC; M. M. Ali, F. Li, Z. Zhang, K. Zhang, D.-K. Kang, J. A. Ankrum, X. C. Le and W. Zhao, Chem. Soc. Rev., 2014, 43, 3324 RSC.
- J. Van Ness, L. K. Van Ness and D. J. Galas, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 4504 CrossRef CAS PubMed; E. Tan, B. Erwin, S. Dames, K. Voelkerding and A. Niemz, Clin. Chem., 2007, 53, 2017 CrossRef; M. Huang, X. Zhou, H. Wang and D. Xing, Anal. Chem., 2018, 90, 2193 CrossRef PubMed; X. Wang, H. Wang, C. Liu, H. Wang and Z. Li, Chem. Commun., 2017, 53, 1124 RSC; J. Nie, D. W. Zhang, F. T. Zhang, F. Yuan, Y. L. Zhou and X. X. Zhang, Chem. Commun., 2014, 50, 6211 RSC; M. S. Reid, X. C. Le and H. Zhang, Angew. Chem., Int. Ed., 2018, 57, 11856 CrossRef PubMed.
- J. Compton, Nature, 1991, 350, 91 CrossRef CAS PubMed; X. Zhao, T. Dong, Z. Yang, N. Pires and N. Høivik, Lab Chip, 2012, 12, 602 RSC; C. M. Mugasa, T. Laurent, G. J. Schoone, P. A. Kager, G. W. Lubega and H. D. F. H. Schallig, J. Clin. Microbiol., 2009, 47, 630 CrossRef PubMed; J. S. Gootenberg, O. O. Abudayyeh, J. W. Lee, P. Essletzbichler, A. J. Dy, J. Joung, V. Verdine, N. Donghia, N. M. Daringer, C. A. Freije, C. Myhrvold, R. P. Bhattacharyya, J. Livny, A. Regev, E. V. Koonin, D. T. Hung, P. C. Sabeti, J. J. Collins and F. Zhang, Science, 2017, 356, 438 CrossRef PubMed.
- M. Vincent, Y. Xu and H. Kong, EMBO Rep., 2004, 5, 795 CrossRef CAS PubMed; E. Torres-Chavolla and E. C. Alocilja, Biosens. Bioelectron., 2011, 26, 4614 CrossRef PubMed.
- O. Piepenburg, C. H. Williams, D. L. Stemple and N. A. Armes, PLoS Biol., 2006, 4, 1115 CrossRef CAS PubMed.
- G. T. Walker, M. C. Little, J. G. Nadeau and D. D. Shank, Proc. Natl. Acad. Sci. U. S. A., 1992, 89, 392 CrossRef CAS PubMed; B. J. Toley, I. Covelli, Y. Belousov, S. Ramachandran, E. Kline, N. Scarr, N. Vermeulen, W. Mahoney, B. R. Lutz and P. Yager, Analyst, 2015, 140, 7540 RSC.
- T. Notomi, H. Okayama, H. Masubuchi, T. Yonekawa, K. Watanabe, N. Amino and T. Hase, Nucleic Acids Res., 2000, 28, E63 CrossRef CAS PubMed; J. Dong, Q. Xu, C. C. Li and C. Y. Zhang, Chem. Commun., 2019, 55, 2457 RSC; C. Ma, F. Wang, X. Wang, L. Han, H. Jing, H. Zhang and C. Shi, Chem. Commun., 2017, 53, 10696 RSC.
- A. R. Connolly and M. Trau, Angew. Chem., Int. Ed., 2010, 49, 2720 CrossRef CAS PubMed; Y. Zhang and C. Y. Zhang, Anal. Chem., 2012, 84, 224 CrossRef PubMed; L. Tian and Y. Weizmann, J. Am. Chem. Soc., 2013, 135, 1661 CrossRef PubMed; J.-F. Huang, N. Zhao, H.-Q. Xu, H. Xia, K. Wei, W.-L. Fu and Q. Huang, Anal. Bioanal. Chem., 2016, 408, 7437 CrossRef PubMed; X. Sun, L. Wang, M. Zhao, C. Zhao and S. Liu, Chem. Commun., 2016, 52, 11108 RSC; C. Shi, Q. Liu, M. Zhou, H. Zhao, T. Yang and C. Ma, Sens. Actuators, B, 2016, 222, 221 CrossRef; Y. Qian, T. Fan, P. Wang, X. Zhang, J. Luo, F. Zhou, Y. Yao, X. Liao, Y. Li and F. Gao, Sens. Actuators, B, 2017, 248, 187 CrossRef; J. S. Gootenberg, O. O. Abudayyeh, M. J. Kellner, J. Joung, J. J. Collins and F. Zhang, Science, 2018, 360, 439 CrossRef PubMed.
- J. Finsterer, H. F. Harbo, J. Baetz, C. Van Broeckhoven, S. Di Donato, B. Fontaine, P. De Jonghe, A. Lossos, T. Lynch, C. Mariotti, L. Schöls, A. Spinazzola, Z. Szolnoki, S. J. Tabrizi, C. M. E. Tallaksen, M. Zeviani, J.-M. Burgunder and T. Gasser, Eur. J. Neurol., 2009, 16, 1255 CrossRef CAS PubMed.
- T. N. Grossman, L. Röglin and O. Seitz, Angew. Chem., Int. Ed., 2007, 46, 5223 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9cc04226a |
| This journal is © The Royal Society of Chemistry 2019 |