
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelf curing and voltage activated catechol adhesives†
Lu
Gan‡
a,
Nigel C. S.
Tan‡
a,
Avi
Gupta
b,
Manisha
Singh
ac,
Oleksandr
Pokholenko
a,
Animesh
Ghosh
a,
Zhonghan
Zhang
a,
Shuzhou
Li
 a and
Terry W. J.
Steele
*a
a and
Terry W. J.
Steele
*a
aSchool of Materials Science and Engineering (MSE), Division of Materials Technology, Nanyang Technological University (NTU), Singapore 639798. E-mail: wjsteele@ntu.edu.sg
bDepartment of Materials Science and Engineering, Indian Institute of Technology, Kanpur, India
cNTU-Northwestern Institute for Nanomedicine (NNIN), Interdisciplinary Graduate School (IGS), Nanyang Technological University (NTU), Singapore 637553
First published on 5th August 2019
Abstract
Catechol adhesives are limited to two-part curing designs. For the first time, a one-pot catechol adhesive is demonstrated with multiple modes of external activation: self-curing, electrocuring, substrate, and two-part curing. Lap shear adhesion (50 kPa) and viscoelastic properties demonstrate that electrocuring is non-inferior to two-part curing methods.
Catechol-grafted polymers/conjugates are promising hydrogels towards bonding of biomaterials on a variety of surfaces.1–3 When catechol is oxidized to o-quinone, the latter covalently reacts with amines or itself by a number of mechanisms.4–6 Although chemical and chelate-mediated crosslinking provides rapid gelation, it requires adding additional initiators that need to be thoroughly mixed. Oxidants,7,8 chelating metal ions,8 and enzymes9 have been demonstrated to initiate the catechol crosslinking, where all fall under the designation of two-part chemical curing (Fig. 1a). Two-part curing has the advantage of rapid gelation, but this suffers from limited manipulation before application, local cytotoxicity, and narrow pH ranges.6,10 Thus, an unmet need exists for a one-pot catechol-mediated adhesive that allows self-curing (with a known lag-time) or through an external stimulus.
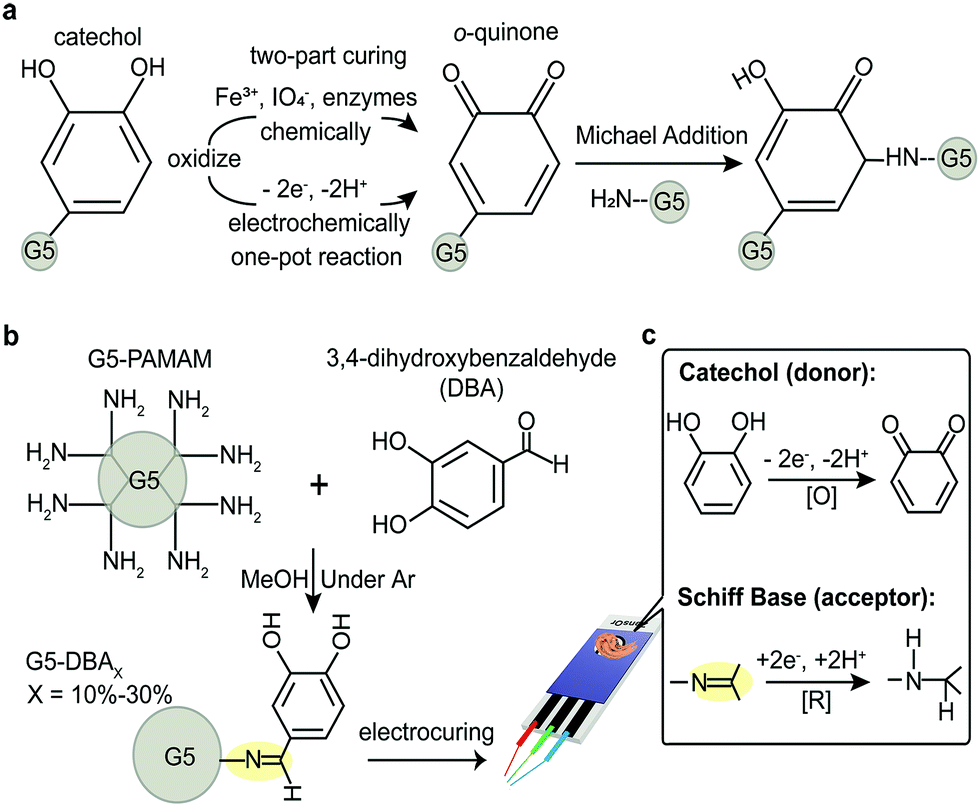 | ||
| Fig. 1 (a) Schematic diagram of catechol being chemically oxidized or electrochemically oxidized into o-quinone, which is responsible to irreversible covalent crosslinking via Michael addition, for example. (b) Synthetic route of 3,4-dihydroxybenzaldehyde (DBA) grafted G5-PAMAM conjugates (G5-DBAX), with the grafting ratio X = 10%, 20%, and 30%, respectively. (c) Schematic illustration of plausible intramolecular donor/acceptor pair in G5-DBAX conjugates, which effects the electrocuring performance. | ||
Low voltage initiation of Voltaglue bioadhesives has recently been demonstrated on matrices that incorporate donor/acceptor pairs.11,12 However, these first proof-of-concept Voltaglue designs required activation of diazirine, whose voltage activation occurs in the range of water electrolysis (−1.6 V vs. −1.23 V, respectively)—imparting significant foaming into the matrix from H2 and O2 evolution.11 Catechols, in comparison, are electrochemically activated well below water electrolysis. Considering the known advantages of catechol redox properties,13 a design of donor/acceptor pair forms our central hypothesis: dendrimer conjugates that graft catechols as electron/proton donor(s) paired with a reducible acceptor(s) will be voltage activated for quinone-mediated crosslinking and adhesion. This design would elevate catechols to one-pot adhesives with material properties that are dependent on electric external stimuli.
Previous work on voltage activated adhesives suggests that donor/acceptor pairs mediate electron transduction, even in a non-aqueous matrix.12 Catechol is oxidized to reactive intermediates, o-quinone, which are responsible for intermolecular crosslinking. Several catechol small molecules are available with handles for dendrimer grafting. A strategy to simultaneously graft catechol (donor) and an acceptor group would reduce the synthesis to a one-pot reaction. Protocatechuic aldehyde or 3,4-dihydroxybenzaldehyde (DBA) is a naturally derived catechol that spontaneously forms Schiff bases (azomethines) in the presence of amines. Schiff bases are regularly exploited for grafting and are rapidly reduced under aqueous conditions with mild reducing agents (e.g., sodium borohydride in SI Results, ESI†).14,15 Simple mixing of DBA with G5-PAMAM (Fig. 1b) grafts both the donor (catechol) and the acceptor (Schiff base) in flawless 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratios to form zwitterionic tautomers that are thermodynamically stable (Fig. S1 and S2, ESI†). G5-PAMAM is utilized as the polymer macromolecule, as it offers numerous design advantages.11,12,16,17 The surface primary amines on the spherical dendrimer limit intramolecular crosslinking while preventing linear entanglements.18,19 It is also soluble in both aqueous and organic solvents, unlike chitosan macromers. PAMAM branched polymer is a model system for exploring structure activity relationships while maintaining an excess of amines for quinone-mediated crosslinking—further modifications are necessary for bioadhesive or underwater applications. Grafting ratios of 10–30% catechol conjugates are explored (G5-DBAX with X = 10%, 20%, 30%, respectively), but ratios >40% had limited aqueous solubility (data not shown).
:1 molar ratios to form zwitterionic tautomers that are thermodynamically stable (Fig. S1 and S2, ESI†). G5-PAMAM is utilized as the polymer macromolecule, as it offers numerous design advantages.11,12,16,17 The surface primary amines on the spherical dendrimer limit intramolecular crosslinking while preventing linear entanglements.18,19 It is also soluble in both aqueous and organic solvents, unlike chitosan macromers. PAMAM branched polymer is a model system for exploring structure activity relationships while maintaining an excess of amines for quinone-mediated crosslinking—further modifications are necessary for bioadhesive or underwater applications. Grafting ratios of 10–30% catechol conjugates are explored (G5-DBAX with X = 10%, 20%, 30%, respectively), but ratios >40% had limited aqueous solubility (data not shown).
Reaction efficiency and final grafting ratios are assessed by 1H NMR (Fig. S3–S6 and Tables S1, S2, ESI†) and size exclusion chromatography (Fig. S7 and Table S2, ESI†). Generation of the Schiff base is complete after 8 h as assessed by UV/vis at 405 nm (Fig. S7, ESI†). This peak is extinguished after borohydride reduction, which removes the Schiff base acceptor as a control (Fig. S8, ESI†). Molar mass of G5-DBAX positively correlates with the grafting ratio with a reduction of the UV/RI peak elution volume (Fig. S7e, ESI†). Redox properties of G5-DBAX conjugates are examined by cyclic voltammetry (CV) in isotonic PBS electrolyte (Fig. 2a and Fig. S9, ESI†). A catechol-free benzaldehyde is also grafted on G5-PAMAM to serve as a catechol-free control (G5-Benz20, Fig. S10, ESI†), and borohydride reduced G5-DBA20 serves as Schiff base free control (reduced G5-DBA20, Fig. S8, ESI†). Free DBA (Fig. S9a, ESI†) exhibits an irreversible and diffusion-controlled redox behavior,20 supported by the following observations: (1) the peak current ratio (IEpa0/IEpc0) is less than 1 at the scan rate of 50 mV s−1; (2) both the anodic peaks (Epa0 = 0.52 V) and the cathodic peaks (Epc0 = −0.02 V) (vs. Ag/AgCl) increase progressively with the increasing scan rates (ν); (3) the plot of peak current as a function of ν1/2 is linear for both peak Epa0 and Epc0 (Fig. S9b, ESI†). The successive CV cycles (either use glassy carbon or Pt as working electrode) exhibited non-repeatable current signal (Fig. S9c and d, ESI†), suggesting that the electrode surface is fouled by electropolymerized DBA film, which in turn inactivates the electrode surface sensitivity.21,22 At the scan rate of 50 mV s−1, the cyclic voltammograms of G5-DBA10 retains the irreversible redox behavior of DBA. G5-DBA10 displays two anodic peaks (Epa1 = 0.29 V and Epa2 = 0.91 V) and a minute cathodic peak (Fig. 2a). Scan rate dependent CV confirms the existence of the Epa2 peak (Fig. S9e, ESI†). The origin of Epa2 peak is attributed to the aldehyde/Schiff base, as it is observed in free DBA, G5-Ben10 (Epa3 in Fig. S9f, ESI†), G5-DBA10, but not in G5-PAMAM (Fig. 2a). All scans of G5-Ben10 are plotted the first cycle (Fig. S9e and f, ESI†) since the peak current decreases after the second cycle. Epa2 results from the known redox activation of Schiff bases under similar conditions.23–27
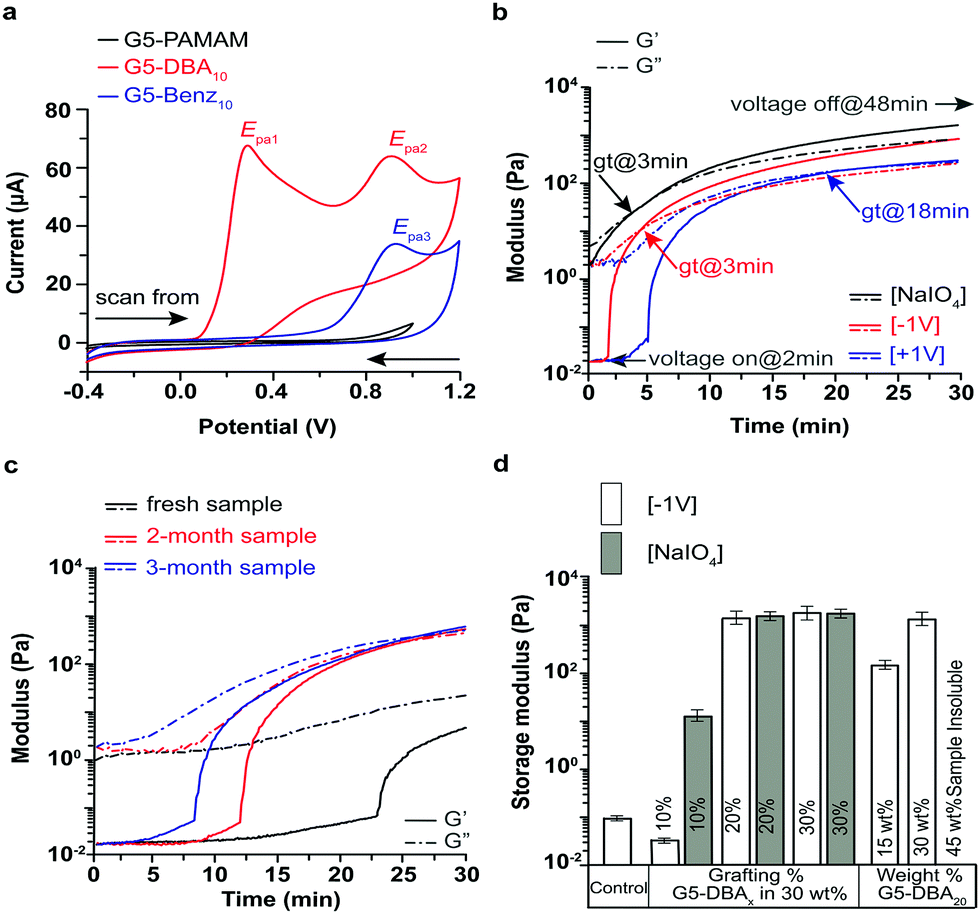 | ||
| Fig. 2 (a) Cyclic voltammograms of G5-PAMAM (as background), G5-Benz10 (as reference), and G5-DBA10 at 50 mV s−1. Electrolyte: PBS (pH 7.2), working electrode: glassy carbon, reference electrode: Ag/AgCl, counter electrode: Pt. Real time dynamic mechanical analysis of formulation of 30 wt% G5-DBA20 conjugates in PBS electrolyte: (b) two-part curing (mixed with 0.1 mM NaIO4 solution) and electrocuring (voltage off: 0–2 min; voltage on: 2–48 min, throughout) at ±1 V, gt—gelation time. (c) self-curing (no voltage applied and no oxidant added) after rehydration. Storage modulus (G′, solid line) and loss modulus (G′′, dot dashed line). (d) G′ values of G5-PAMAM (control) and 30 wt% G5-DBAX formulation as function of grafting % initiated by −1 V voltage or NaIO4 oxidant and G5-DBA20 formulation as function of weight%. 45 wt% formulation is insoluble in PBS. White columns-electrocuring at −1 V, grey columns-two-part curing with periodate. | ||
Real-time rheology is applied to evaluate the mechanical properties (storage modulus-G′ and loss modulus-G′′) before and after curing stimuli. The customized rheology platform incorporates a ceramic probe to avoid catechol–metal chelates, as aluminum probes/surfaces are found to instantly initiate curing (Fig. S10a, ESI†). Voltages of +1 V and −1 V effects on G5-DBAX formulation's viscoelastic properties, gelation (when G′ = G′′), and lag time (period before gelation) are compared to a typical two-part curing method (Fig. 2b). Two-part curing with periodate on G5-DBA20 achieves gelation within 3 min and exhibits a kPa shear modulus after 25 minutes. Negative voltage (cathode as WE) displays instantaneous initiation (Fig. 2b), which is removed after reduction of Schiff base (Fig. S8c, ESI†). Voltage initiation rapidly increases G′, with gelation seen at 2.9 min after voltage activation. At 0 V (control), the G5-DBA20 observes self-curing with a lag-time of 25 ± 5 min following aqueous reconstitution (Fig. 2c). Self-curing is found to be tunable with exposure to aerobic environments in a dry state, where oxygen exposure can increase ratios of catechol/quinone (Fig. S1, ESI†). Rapid initiation of crosslinking (fast increase of G′) is observed at −1 V for 15 and 30 wt% formulations (Fig. 2d). High solute (45 wt%) reconstituted formulations are solid aqueous gels but dissolve in methanol (data not shown). When +1 V is applied, a lag time of 2–3 min is noted before crosslinking with a retarded gelation time of 18 min. The voltage polarity simply swaps the working electrode into a cathode (−1 V, acceptor reduction) or anode (+1 V, donor oxidation), where the working/counter electrodes have a surface area ratio of 4/1, respectively. In the catechol-free G5-Benz20 formulation, no crosslinking is observed at either polarity; therefore, crosslinking is solely mediated by the oxidation of catechol (Fig. S10, ESI†). However, the Schiff base-free reduced G5-DBA20 loses the ability to be instantly reduced and also has a delayed gelation time of 8.4 min vs. 3 min. Taken together, these observations suggest that reduction of the Schiff base is the rate limiting reaction under the −1 V conditions, whereby a cathode working electrode facilitates rapid initiation by providing a larger area. Oxygen mediated pathways are responsible for the self-curing behavior, and the +1 V is likely a mixture of both mechanisms. However, minimum grafting of catechol is required before voltage mediated crosslinking is observed, as G5-DBA10 in Fig. S10 (ESI†) displays no gelation within 30 min but can be chemically cured. Fig. 2d compares the G′ values of all electrocuring formulations to two-part curing with periodate. G5-DBA20 and G5-DBA30 activation by electrocuring or periodate displayed no significant differences after 60 min.
Lap shear adhesion is evaluated with wet collagen films, which serve as a mimic of wet tissue substrates.28 G5-DBA20 at 30 wt% is applied to the collagen and activated with disposable 3-electrode chips (Fig. 3). G5-DBA20 formulation is cured by two methods: electrocuring (−1 V) vs. two-part curing (periodate). Electrocuring at +1 V is not evaluated due to the inferior material properties and gelation time, as shown in the electrorheology analyses. Lap shear adhesion strength at failure evaluates the crosslinked matrix since cohesive failure is seen throughout.
 | ||
| Fig. 3 Schematic illustration of: (a) electrocuring of 30 wt% G5-DBA20 formulation with the 3-electrode chip and wet collagen film. Inset: Geometry and effective working area of electrodes on Zensor®chip. WE: working electrode, CE: counter electrode, RE: reference electrode. FN indicates the pulling force and directions. (b) Representative stress/strain plots of 30 wt% G5-DBA20 formulations: self-curing, electrocuring (−1 V), and two-part curing (with 0.1 mM NaIO4). (c) Lap shear adhesion strength of 30 wt% G5-DBA20 formulation in comparison of self-curing, electrocuring, and two-part curing in <30 min and >60 min curing period. | ||
Tack evaluation under 30 min demonstrates that electrocuring G5-DBA20 formulation has a significant increase over two-part curing, and no adhesion is present for the self-curing formulation. The self-curing control sample (atmosphere exposure with neither voltage nor periodate applied) displays only viscous liquid material properties (Fig. 3b). The lap shear adhesion strength comparisons after 60 min are indicated in Fig. 3c. The self-curing formulation has an increase of adhesion strength to ∼4 N cm−2 (40 kPa), which is about 4 times higher than 30 min case. Electrocuring continuously for 60 min is non-inferior to two-part curing, which both present ∼5 N cm−2 (50 kPa) adhesion strength. In summary, an azomethine/catechol adhesive is simply synthesized under ambient conditions to yield an acceptor/donor pair. The spontaneous reaction creates a redox-responsive adhesive that can be applied and activated by a number of external stimuli. The inclusion of both electron donor and acceptor groups creates an adhesive that can self-cure after a predictable lag-time. Activation can be triggered under both oxidative and reducing environments. For example, gelation time decreased in the following oxidative conditions: ambient atmosphere, +1 V, or periodate. A reducing environment triggered cross-linking instantaneously, −1 V or aluminum substrates (Al → Al3+ + 3e−). A serendipitous finding was the short-term stability in aqueous solvents, but this allows development of self-curing adhesion that is semi-stable in a dry state. A bioadhesive that allows liquid manipulation and then self-cures with a known lag-time is a current unmet need for tissue repair. Activation of the lag-time ‘clock’ is as simple as aqueous reconstitution. The lag time of the dried and precipitated formulation drifted over a period of 3 months when exposed to aerobic environments. This suggests that the lag-time may be optimized in oxygen-free or limited exposure conditions (Fig. 2c). Future work will address tuning lag-time and further shelf-life stability.
For the first time, a catechol-adhesive allows activation by electrocuring, instead of traditional two-part curing method. This allows activation while avoiding side-effects from oxidation agents or highly concentrated metal chelators. Electrocuring adhesive strength was comparable to the periodate two-part curing methods. Two-part curing is a standard method of activation of mussel biomimetic adhesives, with modest adhesion strength 40 kPa.8 Polydopamine-co-acrylate cured with periodate achieved up to 70 kPa after one day of curing. Electrocuring allows a simpler approach with no additive mixing and more precise control over initiation and gelation time. However, formulations appear to be limited in design—successful electrocuring was only observed at 20–30% grafting ratios. Too little (10%) grafting prevented voltage-activation—too much (greater than 40%) displayed aqueous solubility problems that may stem from spontaneous crosslinking under storage. The electrocuring formulations herein have some advantages over our previous Voltaglue formulations.11,12,29 This redox donor/acceptor adhesives could be activated at ±1 V (vs. Ag/AgCl), but Voltaglue formulations need a higher voltage of −1.6 V that evolves gases which can ultimately weaken the adhesive matrix and limit strength. However, Voltaglue appears more stable in oxidative and aqueous environments, as no spontaneous self-curing has been observed under ambient conditions.
To our surprise, voltage-activated crosslinking was accelerated when the working electrode was set as the cathode, effectively providing a larger surface area for reductive reactions. This supports the proposed hypothetical acceptor/donor mechanism in Fig. 1, but no direct evidence exists of Schiff base reduction. This is likely due to a combination of factors; G5-PAMAM contains both primary and tertiary amines, providing a local alkaline environment (pH 9–10). If zwitterionic, the Schiff base catechol would attract electrons to the protonated azomethine (Fig. S1, ESI†). At this high pH, protons are limited to the zwitterion or other tautomers known to exist under aqueous conditions (see SI Results, ESI†). A −1 V electrochemical gradient at the working electrode attracts protons from atmosphere O2-mediated oxidation of catechol, which is thermodynamically favorable (but the internal electronic reduction/oxidation is not, see Fig. S2, ESI†). With Schiff bases speculated to be reduced at this −1 V potential, catechol to quinone oxidations are uninhibited. As quinones are responsible for crosslinking, their formation will decrease the time to gelation. Supporting this hypothesis is the empirical result that the aryl-Schiff bases are reduced with sodium borohydride (Fig. S8, ESI†), where borohydride anion is estimated to have a formal potential of E°′ = −0.7 to −0.43 V.30
Many formulations of chitosan–catechol conjugates have been synthesized—chitosan is a cheap and scalable branched polymer that has been incorporated in many biomaterials. However, chitosan has an inherent solubility limit in acidic mediums that is often addressed with additional grafting. Even with catechol grafting, the functionalized chitosan can only achieve a 6 wt% solution (polymer in aqueous solvent)—which is too dilute for voltage-activation (G5-DBA20 at 10 wt% displays no electrocuring).6,31 Grafting 19% to 80% of the total amines with catechol on chitosan displays similar material properties as the reported G5-DBA20 formulations.15,32 Others have exploited electrochemical synthesis of chitosan–catechol, where reductive amination grafts chitosan to the surface followed by oxidative activation for catechol–chitosan grafting towards anti-oxidant surfaces.33 The technology herein may allow a more straightforward approach for similar purposes. G4-PAMAM was grafted with PEG–catechol to form high modulus hydrogels up to 80 kPa,34 but this required two-part curing with periodate and cure times of 48 hours. Future work will address methods to achieve similar high moduli with reversible, self-curing, and electrocuring properties while building on prior work to achieve in vivo implants and blood biocompatibility.35–37
Conflicts of interest
There are no conflicts to declare.Notes and references
- H. Lee, S. M. Dellatore, W. M. Miller and P. B. Messersmith, Science, 2007, 318, 426–430 CrossRef CAS PubMed.
- J. Yu, W. Wei, M. S. Menyo, A. Masic, J. H. Waite and J. N. Israelachvili, Biomacromolecules, 2013, 14, 1072–1077 CrossRef CAS PubMed.
- J. Y. Park, J. S. Kim and Y. S. Nam, Carbohydr. Polym., 2013, 97, 753–757 CrossRef CAS PubMed.
- M. Mehdizadeh, H. Weng, D. Gyawali, L. Tang and J. Yang, Biomaterials, 2012, 33, 7972–7983 CrossRef CAS PubMed.
- J. Yang, M. A. Cohen Stuart and M. Kamperman, Chem. Soc. Rev., 2014, 43, 8271–8298 RSC.
- J. H. Ryu, S. Hong and H. Lee, Acta Biomater., 2015, 27, 101–115 CrossRef CAS PubMed.
- G. Westwood, T. N. Horton and J. J. Wilker, Macromolecules, 2007, 40, 3960–3964 CrossRef CAS.
- B. J. Kim, D. X. Oh, S. Kim, J. H. Seo, D. S. Hwang, A. Masic, D. K. Han and H. J. Cha, Biomacromolecules, 2014, 15, 1579–1585 CrossRef CAS PubMed.
- J. H. Ryu, Y. Lee, M. J. Do, S. D. Jo, J. S. Kim, B.-S. Kim, G.-I. Im, T. G. Park and H. Lee, Acta Biomater., 2014, 10, 224–233 CrossRef CAS PubMed.
- M. Cencer, Y. Liu, A. Winter, M. Murley, H. Meng and B. P. Lee, Biomacromolecules, 2014, 15, 2861–2869 CrossRef CAS PubMed.
- J. Ping, F. Gao, J. L. Chen, R. D. Webster and T. W. J. Steele, Nat. Commun., 2015, 6, 8050 CrossRef CAS PubMed.
- L. Gan, N. C. S. Tan, A. H. Shah, R. D. Webster, S. L. Gan and T. W. J. Steele, Macromolecules, 2018, 51, 6661–6672 CrossRef CAS.
- D. Nematollahi and S. Dehdashtian, Tetrahedron Lett., 2008, 49, 645–649 CrossRef CAS.
- J. H. Billman and A. C. Diesing, J. Org. Chem., 1957, 22, 1068–1070 CrossRef CAS.
- P. S. Yavvari and A. Srivastava, J. Mater. Chem. B, 2015, 3, 899–910 RSC.
- H. S. Nanda, M. Singh and T. W. J. Steele, ECS Trans., 2017, 77, 547–555 CrossRef CAS.
- L. Gan, N. C. S. Tan and T. W. J. Steele, ECS Trans., 2017, 77, 981–988 CrossRef CAS.
- P. K. Maiti, T. Çaǧın, G. Wang and W. A. Goddard, Macromolecules, 2004, 37, 6236–6254 CrossRef CAS.
- C. Ornelas, Macromol. Chem. Phys., 2016, 217, 149–174 CrossRef CAS.
- A. J. Bard and L. R. Faulkner, Electrochemical Methods: Fundamentals and Applications, Wiley, 2000 Search PubMed.
- D. Nematollahi and S. M. Golabi, Electroanalysis, 2001, 13, 1008–1015 CrossRef CAS.
- M. F. Delbem, W. J. Baader and S. H. P. Serrano, Quim. Nova, 2002, 25, 741–747 CrossRef CAS.
- H. Ohmori, A. Matsumoto, M. Masui and H. Sayo, J. Electrochem. Soc., 1977, 124, 1849–1854 CrossRef CAS.
- M. Masui and H. Ohmori, J. Chem. Soc., Perkin Trans. 2, 1972, 1882–1887 RSC.
- C. I. Simionescu, M. Grovu-Ivanoiu, I. Cianga, M. Grigoras, A. Duca and I. Cocârla, Angew. Makromol. Chem., 1996, 239, 1–12 CrossRef CAS.
- R. Das, A. Saxena, S. Saxena and G. Khan, J. Adv. Electrochem., 2015, 1, 19–24 Search PubMed.
- E. H. Cordes and W. P. Jencks, J. Am. Chem. Soc., 1963, 85, 2843–2848 CrossRef CAS.
- T. W. J. Steele, C. L. Huang, E. Nguyen, U. Sarig, S. Kumar, E. Widjaja, J. S. C. Loo, M. Machluf, F. Boey, Z. Vukadinovic, A. Hilfiker and S. S. Venkatraman, J. Mater. Sci.: Mater. Med., 2013, 24, 2013–2027 CrossRef CAS PubMed.
- M. Singh, H. S. Nanda, R. D. O'Rorke, A. E. Jakus, A. H. Shah, R. N. Shah, R. D. Webster and T. W. J. Steele, Adv. Healthcare Mater., 2018, 7, 1800538 CrossRef PubMed.
- N. G. Connelly and W. E. Geiger, Chem. Rev., 1996, 96, 877–910 CrossRef CAS PubMed.
- K. Kim, J. H. Ryu, D. Y. Lee and H. Lee, Biomater. Sci., 2013, 1, 783–790 RSC.
- J. Xu, S. Strandman, J. X. X. Zhu, J. Barralet and M. Cerruti, Biomaterials, 2015, 37, 395–404 CrossRef CAS PubMed.
- C. Cao, E. Kim, Y. Liu, M. Kang, J. Li, J.-J. Yin, H. Liu, X. Qu, C. Liu, W. E. Bentley and G. F. Payne, Biomacromolecules, 2018, 19, 3502–3514 CrossRef PubMed.
- Y. Wang, Q. Zhao, Y. Luo, Z. Xu, H. Zhang, S. Yang, Y. Wei and X. Jia, Chem. Commun., 2015, 51, 16786–16789 RSC.
- H. S. Nanda, A. H. Shah, G. Wicaksono, O. Pokholenko, F. Gao, I. Djordjevic and T. W. J. Steele, Biomacromolecules, 2018, 19, 1425–1434 CrossRef CAS PubMed.
- F. Gao, I. Djordjevic, O. Pokholenko, H. Zhang, J. Zhang and T. W. J. Steele, Molecules, 2018, 23, 796 CrossRef PubMed.
- A. H. Shah, O. Pokholenko, H. S. Nanda and T. W. J. Steele, Mater. Sci. Eng., C, 2019, 100, 215–225 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9cc04166d |
| ‡ These authors contributed equally to the work. |
| This journal is © The Royal Society of Chemistry 2019 |