
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Flavonoid–alkylphospholipid conjugates elicit dual inhibition of cancer cell growth and lipid accumulation†
Zhengwei
Zhou‡
a,
Biyao
Luo‡
a,
Xi
Liu‡
a,
Mimi
Chen
a,
Wenjun
Lan
bc,
Juan L.
Iovanna
 c,
Ling
Peng
*b and
Yi
Xia
*a
c,
Ling
Peng
*b and
Yi
Xia
*a
aChongqing Key Laboratory of Natural Product Synthesis and Drug Research, School of Pharmaceutical Sciences, Chongqing University, China. E-mail: yixia@cqu.edu.cn
bAix-Marseille Université, CNRS, Centre Interdisciplinaire de Nanoscience de Marseille, Equipe Labellisé par La Ligue, France. E-mail: ling.peng@univ-amu.fr
cCentre de Recherche en Cancérologie de Marseille (CRCM), INSERM U1068, CNRS, Aix-Marseille Université and Institut Paoli-Calmettes, Marseille 13288, France
First published on 4th July 2019
Abstract
Cancer development is often associated with lipid metabolic reprogramming, including aberrant lipid accumulation. We create novel paradigms endowed with dual functions of anticancer activity and inhibition of lipid accumulation by conjugating the natural product quercetin and synthetic alkylphospholipid drugs, and harnessing the biomedical effects of both. These conjugates offer fresh perspectives in the search for anticancer candidates.
Cancer is a serious disease which causes a major global health burden.1 Cancer development is often associated with genetic and environmental factors; the latter includes obesity, diet, smoking, infections etc.2 For example, individuals who are obese or overweight are at high risk of developing cancer.3 This is because elevated lipid levels, caused by enhanced lipid synthesis and aberrant lipid accumulation, are able to induce carcinogenesis and accelerate metastasis in cancers.4 Therefore, lipid metabolism, including lipid synthesis, transportation, intake and aberrant lipid accumulation, constitutes a promising therapeutic target for treating cancer,5 and suppressing the abnormal accumulation of lipids can be beneficial for cancer treatment.4
Quercetin (Fig. 1) is a natural polyphenol flavonoid well known for its beneficial effects as a phytochemical remedy for many diseases such as obesity, diabetes, inflammation, aging etc.6–8 By virtue of its polyphenol character, quercetin can quench free radicals and therefore possesses strong antioxidant activity.9 Moreover, quercetin exhibits anticancer activity through its antioxidative effect9 or by targeting proteins including fatty acid synthase,10 heat shock proteins11 and various kinases involved in intracellular signaling.12 It should be noted that although quercetin has potent biological activities, it is poorly soluble in water or organic solvents, and hence has poor bioavailability. This has largely limited its therapeutic applications. It is therefore crucial to synthesize novel quercetin derivatives with improved bioavailability as well as enhanced biological activity.
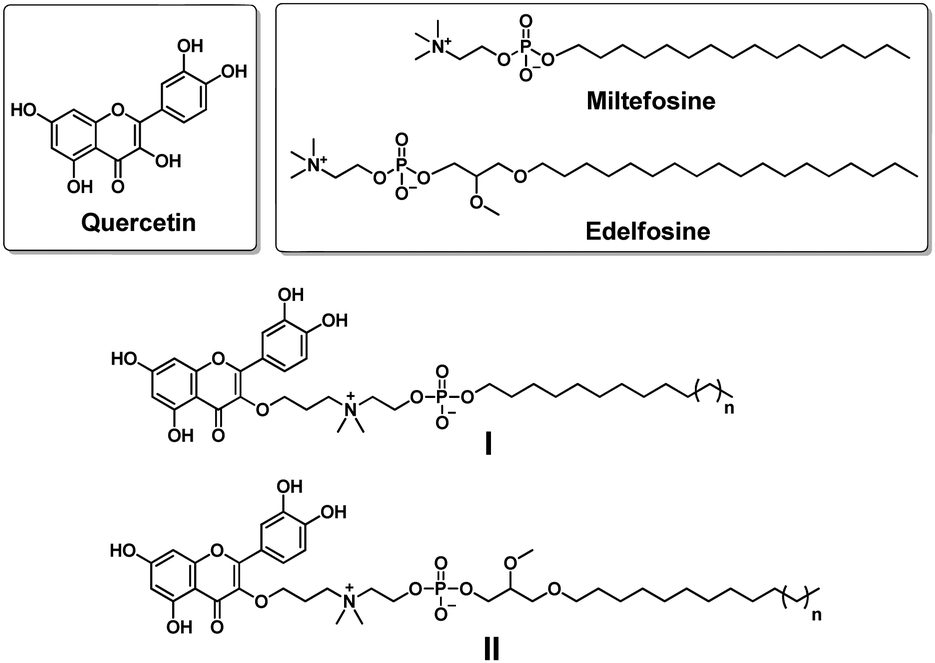 | ||
| Fig. 1 Chemical structures of the natural product quercetin, the synthetic alkylphospholipid drugs miltefosine and edelfosine, and their conjugates I and II studied in this work. | ||
Synthetic alkylphospholipids (APLs) are a class of lipid derivatives which act at the cell membrane to interfere with lipid metabolism and signalling pathways. Several APLs are currently in clinical use, such as miltefosine, edelfosine and perifosine, to treat various diseases (Fig. 1).13,14 APLs structurally resemble the natural lipid lysophosphatidylcholine (LysoPC) and share common features including a long hydrocarbon chain and a phosphocholine polar head group. In addition, they are devoid of fatty acid ester functions, and hence are resistant to catabolic degradation by lipase. Miltefosine is active against various parasite species13 and pathogenic microorganisms (bacteria and fungi),15 and also inhibits cancer cell proliferation,13 whereas edelfosine can induce apoptosis in various malignant cells through targeting lipid rafts and the endoplasmic reticulum.16
Motivated by these considerations, we aimed to conjugate quercetin with APLs with the aim of combining their individual biomedical features, thus generating novel multifunctional drug candidates which possess anticancer activity and affect lipid metabolism. Here, we report our design and synthesis of a series of quercetin–APL conjugates I and II (Fig. 1). We also evaluated these conjugates for their anticancer activity related to their ability to target heat shock proteins and affect lipid metabolism.
In order to provide a robust conjugation while also attempting to improve the bioavailability of quercetin, we decided to bridge quercetin with the N-terminus of the choline moiety in APLs. The synthesis of conjugate I was started with compound I1 (Scheme 1 and Table S1, ESI†), which was prepared by adapting a previously reported method.17I1 was then treated with 1,3-dibromopropane to generate the quaternary ammonium I2. By using a large excess of 1,3-dibromopropane, we successfully suppressed the formation of the dialkylated quaternary ammonium product and readily obtained the mono alkylated quaternary ammonium I2 in good yields. The obtained I2 was condensed with benzyl-protected quercetin to deliver I3. The subsequent deprotection of the benzyl group in I3 using H2/Pd/C afforded the quercetin–APL conjugate I quantitatively (Scheme 1 and Table S1, ESI†). Similarly, conjugate II was also successfully obtained (Scheme 1 and Table S1, ESI†).
 | ||
| Scheme 1 Synthesis of quercetin–alkylphospholipid conjugates I and II. | ||
It is worth noting that unlike quercetin, all the conjugates are soluble in the mixed organic solvent MeOH/CH2Cl2. Although neither I nor II is soluble in water, addition of the detergent sodium dodecylsulfate markedly increased their solubility in phosphate buffer (Fig. S1, ESI†). This demonstrates that the quercetin–APL conjugates retained the amphiphilic properties of APLs and have the potential to improve the bioavailability of quercetin.
We next evaluated the anticancer activity of the synthesized conjugates by assessing their effect on the viability of human liver cancer HepG2 and Hep3B cells, pancreatic cancer Panc-1 cells and ovarian cancer SKOV3 cells using the MTT assay (Table S2, ESI†). Remarkably, the length of the hydrophobic alkyl chain in both I and II highly impacted the biological activity. For both series, the conjugates bearing short alkyl chains (≤C12) lacked the ability to kill cancer cells, whereas those with longer alkyl chains (≥C14) were effective in inhibiting cancer cell growth. In addition, the inhibitory activity was enhanced as the alkyl chain length increased, and the best activity occurred with the C18 alkyl chain in series I (Id in Fig. 2A and Table S2, ESI†), and with C16 in series II (IIc in Fig. 2A and Table S2, ESI†). Both Id and IIc outperformed the parent compounds quercetin, miltefosine and edelfosine against all tested cancer cell lines. It should be mentioned that both compounds I3 and II3 with a protected quercetin motif, or the protected quercetin alone, were inactive (data not shown). This suggests that both the quercetin and the APL structural motifs are critical for imparting anticancer activity to the conjugates.
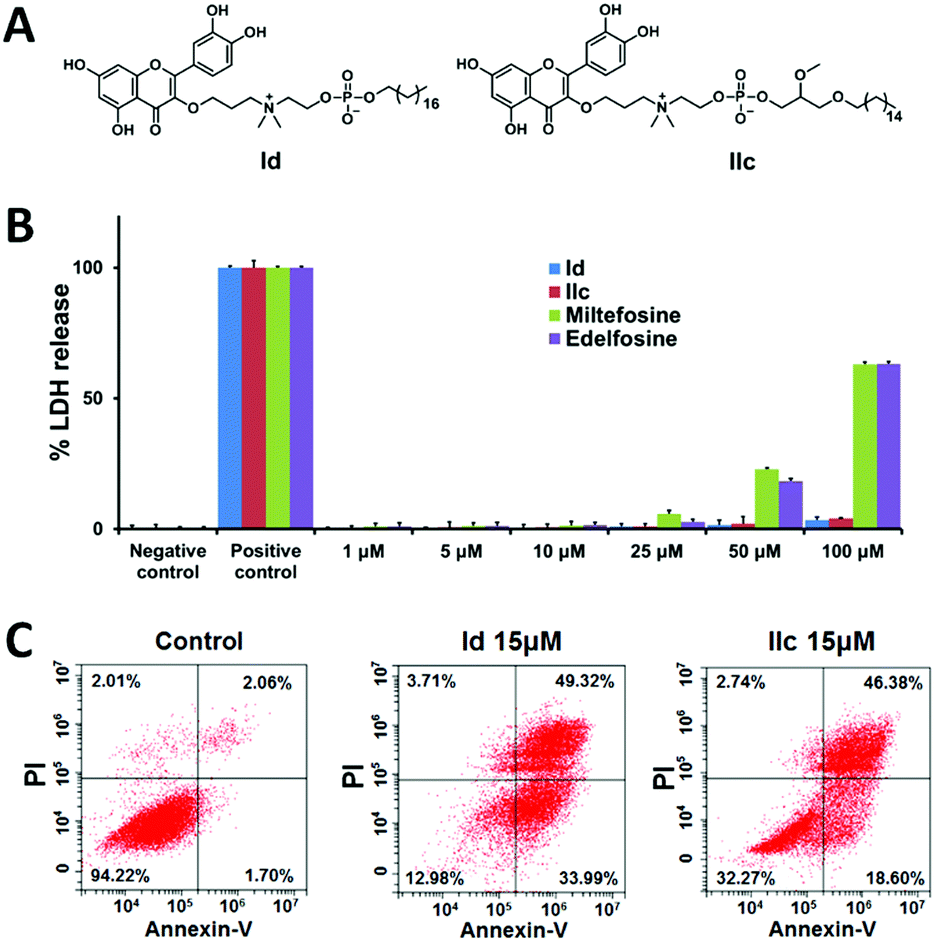 | ||
| Fig. 2 The most active conjugates Id and IIc induce apoptosis in cancer cells, with negligible necrosis. (A) Chemical structures of Id and IIc. (B) LDH assay for necrosis induced by Id, IIc, miltefosine and edelfosine in HepG2 cells after 24 h treatment. The negative control determines the LDH activity released from untreated cells, and the positive control determines the maximum releasable LDH activity in cells treated with lysis buffer. (C) Apoptosis, rather than necrosis, was predominantly induced in HepG2 cells after 48 h treatment with Id and IIc. Flow cytometry was used to quantify the cells undergoing apoptosis (annexin V-positive cells) and necrosis (PI-positive cells). Untreated HepG2 cells were used as the reference control. | ||
We further investigated the most effective compounds Id and IIc in order to understand the mechanism underlying their antiproliferative activity on cancer cells. We suspected that the long hydrophobic alkyl chain and the amphiphilic feature of the quercetin–APL conjugates might induce cell necrosis due to the interaction with the cell membrane. We therefore performed the lactate dehydrogenase (LDH) assay in HepG2 cells to assess the eventual membrane damage induced by Id and IIc. Gratifyingly, no significant induction of LDH release was observed even at concentrations up to 100 μM (Fig. 2B). In contrast, miltefosine and edelfosine began to induce considerable LDH release even at 25 μM. We next examined whether the antiproliferative activity of these conjugates on cancer cells could be ascribed to apoptosis. To do this, we treated the cancer cells with compounds Id and IIc, then stained them with annexin V and propidium iodide (PI) to determine the percentage of apoptotic and necrotic cells using flow cytometric analysis. A remarkable increase in the percentage of apoptotic cells, but only a very small increase in the percentage of necrotic cells, was observed following treatment with Id and IIc (Fig. 2C). Collectively, these results indicate that both Id and IIc induce apoptosis rather than necrosis as the predominant mechanism of cancer cell death.
The anti-apoptotic protein Bcl-2 plays a crucial role in regulating cell death by sequestering pro-apoptotic molecules.18 The cleavage of PARP, a 116 kDa nuclear poly(ADP-ribose) polymerase, is the hallmark of apoptosis. We therefore checked the protein level of Bcl-2 and PARP in HepG2 and Panc-1 cells using western blotting. As we can see in Fig. 3, both compounds Id and IIc effectively reduced the expression of Bcl-2 and facilitated PARP cleavage, consistent with their ability to induce apoptosis.
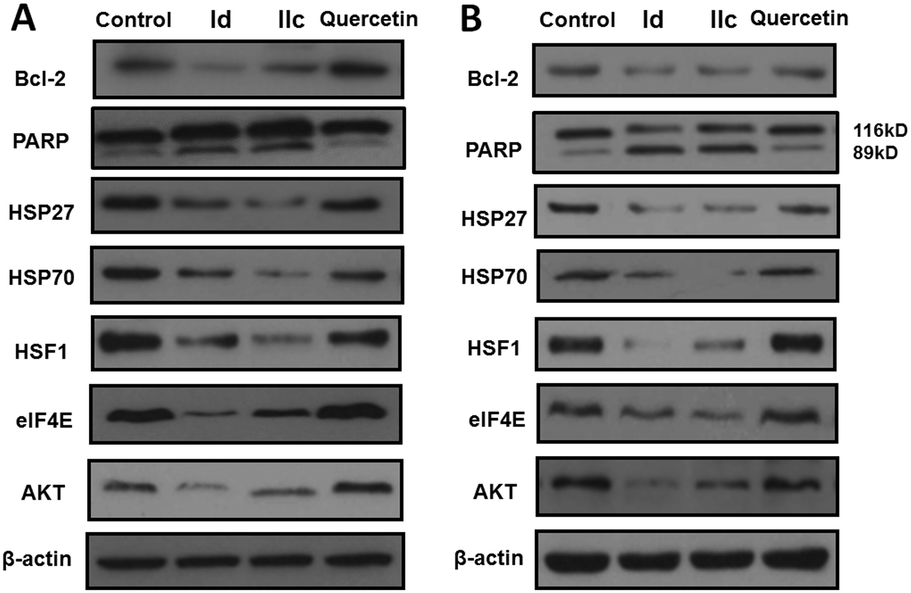 | ||
| Fig. 3 Compounds Id and IIc not only efficiently down-regulate Bcl-2, and facilitate PARP cleavage but also strongly inhibit the expression of HSF1, the downstream heat-shock proteins HSP27 and HSP70, and their client oncoproteins eIF4E and AKT in (A) HepG2 and (B) Panc-1 cells. The Bcl-2, PARP, HSF1, HSP70, HSP27, eIF4E and AKT protein levels after treatment with Id (15 μM), IIc (15 μM) and quercetin (15 μM) were analyzed using western blotting, with β-actin as the reference. | ||
It is known that quercetin executes its anticancer function by inhibiting the induction of several heat shock proteins (HSPs).11 HSPs are a superfamily of molecular chaperones which help proteins to adopt their normal functional architecture. Importantly, HSPs are attractive anticancer targets thanks to their high expression in a plethora of human cancer cells and their function as master regulators of multiple oncoproteins.19 We hence examined the effect of Id and IIc on HSPs with the aim of defining whether they affect the common heat shock response (HSR) pathway. As shown in Fig. 3, the stress-inducible HSP27 and HSP70 were almost fully repressed upon treatment with Id and IIc, while quercetin only slightly inhibited these two proteins. Remarkably, the expression of the primary transcription factor of HSPs, heat shock factor 1 (HSF1),20 was also greatly attenuated upon treatment with Id and IIc, but was unaffected by quercetin (Fig. 3). Therefore, the quercetin–APL conjugates not only enhance the ability of quercetin to target HSPs, but also strongly down-regulate HSF1. These results highlight that Id and IIc potentially regulate the HSR pathway.
As HSPs act as molecular chaperones to stabilize the conformation of their client proteins, the suppression of HSPs causes the degradation and depletion of the corresponding client proteins.19 Eukaryotic translation initiation factor 4E (eIF4E) is a client oncoprotein of HSP27 and has elevated expression in several cancers,21 while AKT, a serine-threonine protein kinase associated with malignancy, is also chaperoned by HSPs.19,22 As expected, both Id and IIc effectively reduced the expression level of eIF4E and AKT, consistent with our observations for HSP27 and HSP70 (Fig. 3). Considering the role of elF4E and AKT in cancer development, the down-regulation of elF4E and AKT could also contribute to the observed anticancer activity of these quercetin–APL conjugates.
Quercetin displays anti-obesity activity in mice and is known to reduce lipid accumulation by modulating lipid metabolism.23 We therefore asked whether the quercetin–APL conjugates could also affect lipid accumulation. As the liver is an important organ for maintaining lipid and glucose homeostasis, we focused our study of the lipid-lowering effect on HepG2 liver cancer cells which have high triglyceride (TG) levels. As shown in Fig. 4A, the number of lipid droplets for lipid deposition in HepG2 cells was greatly reduced following treatment with Id and IIc, while quercetin had a much weaker effect. We further analyzed the cells for the relative TG level, as TG is one of the main constituents of body fat in mammals. Our results in Fig. 4B showed that both Id and IIc significantly decreased the TG content in a dose-dependent manner, whereas quercetin only slightly decreased the TG content, and miltefosine and edelfosine had no notable impact on the TG content. Additionally, it was reported that APLs can inhibit AKT via depletion of lipids in the plasma membrane.24 Therefore, the observed down-regulation of AKT by our synthesized conjugates (Fig. 3) could potentially be ascribed not only to the inhibition of HSPs, but also to lipid depletion.
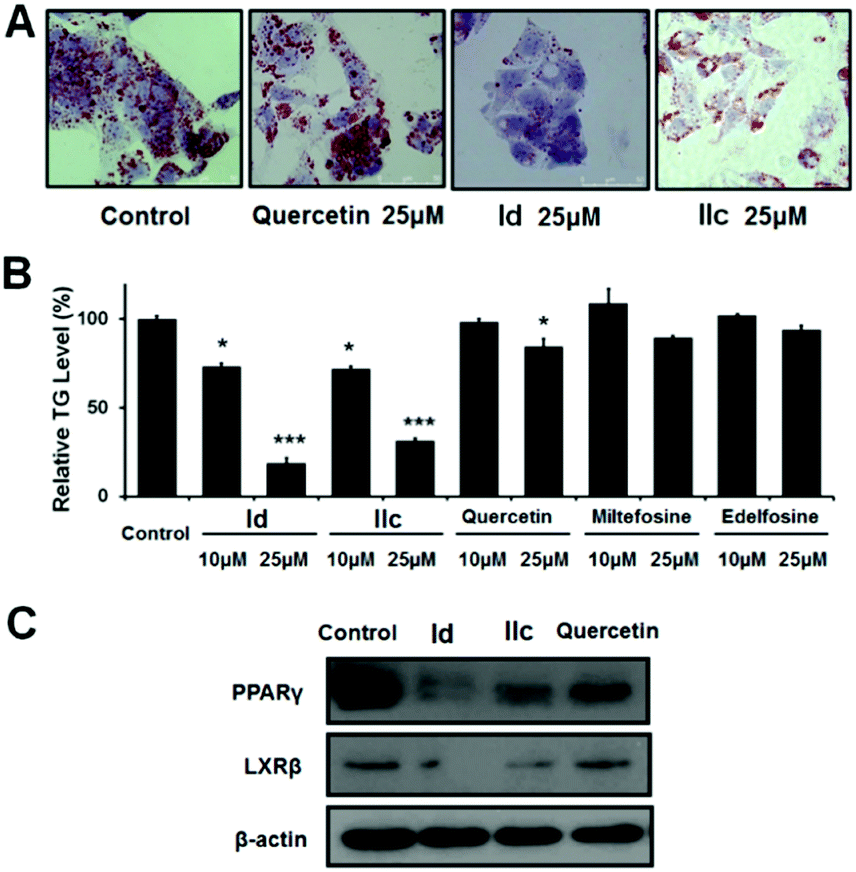 | ||
| Fig. 4 Compounds Id and IIc inhibit lipid accumulation and de novo lipid synthesis in HepG2 cells. (A) Representative images of cells stained with Oil Red O (magnification, 630×). The red dots are lipid droplets in cells. (B) Intracellular triglyceride (TG) content was measured using an enzymatic colorimetric assay and normalized to protein content. Cells were treated with Id, IIc, quercetin, miltefosine and edelfosine. The relative TG level, compared with that of untreated control cells, is expressed as the mean ± S.E. (n = 3). *, p < 0.05; ***, p < 0.001. (C) The level of PPARγ and LXRβ, two proteins related to de novo lipid synthesis, upon treatment with Id (15 μM), IIc (15 μM) and quercetin (15 μM) was analyzed using western blotting, with β-actin as the reference. | ||
The ability of quercetin–APL conjugates to inhibit lipid accumulation suggested that these compounds may regulate de novo lipid synthesis, also referred to as adipogenesis and/or lipogenesis.25,26 Adipogenesis is finely controlled by some key transcription factors or markers of adipocyte differentiation and triglyceride accumulation such as the liver X receptors (LXRs)27 and peroxisome proliferator-activated receptors (PPARs).28 We therefore investigated the protein expression of LXRβ and PPARγ in HepG2 cells using western blotting. Our results showed that Id and IIc were effective in repressing both LXRβ and PPARγ (Fig. 4C). This suggests that Id and IIc may down-regulate the activity of adipocytes, the excess differentiation of which can lead to lipid accumulation, and hence to an increased risk of diabetes and obesity.29 Moreover, targeting LXRβ and PPARγ has been reported as a promising therapeutic approach to modulate tumor progression.30,31 Taken together, our results show that the development of quercetin–APL conjugates may provide a new strategy for generating drug candidates against cancer and other diseases related to abnormal lipid accumulation.
In conclusion, we designed and synthesized a series of dual-function conjugates based on the natural product quercetin and the synthetic alkylphospholipid (APL) drugs miltefosine and edelfosine. These conjugates showed effective activity against the growth of cancer cells, with Id and IIc being the most effective. Notably, they induced apoptosis via the down-regulation of HSF1 together with its downstream heat shock proteins Hsp27 and Hsp70 as well as the anti-apoptotic protein Bcl-2. In addition, both Id and IIc effectively inhibited de novo lipid synthesis and lipid accumulation by lowering the intracellular triglyceride content and suppressing the expression of PPARγ and LXRβ. These results collectively suggest that the quercetin–APL conjugates combine the advantageous characteristics of quercetin and APL drugs, and are endowed with dual biological effects of inhibiting cancer cell growth and lipid synthesis/accumulation. Consequently, these conjugates constitute a promising structural paradigm in the discovery of new anticancer candidates. This study may also open new avenues for developing therapeutic agents to treat other lipid accumulation-related diseases such as obesity, diabetes and cardiovascular diseases. We are actively working in this direction.
This work is supported by the National Natural Science Foundation of China (81502920), the Fundamental Research Funds for the Central University (2018CDYJSY0055) and PHC XU GUANGQI (38748ZF). We thank Dr Alice Carrier for her critical review and helpful comments.
Conflicts of interest
There are no conflicts to declare.Notes and references
- F. Bray, J. Ferlay, I. Soerjomataram, R. L. Siegel, L. A. Torre and A. Jemal, Ca-Cancer J. Clin., 2018, 68, 394–424 CrossRef.
- F. Islami, A. Goding Sauer, K. D. Miller, R. L. Siegel, S. A. Fedewa, E. J. Jacobs, M. L. McCullough, A. V. Patel, J. Ma, I. Soerjomataram, W. D. Flanders, O. W. Brawley, S. M. Gapstur and A. Jemal, Ca-Cancer J. Clin., 2018, 68, 31–54 CrossRef PubMed.
- E. J. Gallagher and D. LeRoith, Physiol. Rev., 2015, 95, 727–748 CrossRef CAS PubMed.
- J. Long, C. J. Zhang, N. Zhu, K. Du, Y. F. Yin, X. Tan, D. F. Liao and L. Qin, Am. J. Cancer Res., 2018, 8, 778–791 Search PubMed.
- Q. Liu, Q. Luo, A. Halim and G. Song, Cancer Lett., 2017, 401, 39–45 CrossRef CAS PubMed.
- G. D'Andrea, Fitoterapia, 2015, 106, 256–271 CrossRef PubMed.
- S. L. Miles, M. McFarland and R. M. Niles, Nutr. Rev., 2014, 72, 720–734 CrossRef.
- M. Xu, T. Pirtskhalava, J. N. Farr, B. M. Weigand, A. K. Palmer, M. M. Weivoda, C. L. Inman, M. B. Ogrodnik, C. M. Hachfeld, D. G. Fraser, J. L. Onken, K. O. Johnson, G. C. Verzosa, L. G. P. Langhi, M. Weigl, N. Giorgadze, N. K. LeBrasseur, J. D. Miller, D. Jurk, R. J. Singh, D. B. Allison, K. Ejima, G. B. Hubbard, Y. Ikeno, H. Cubro, V. D. Garovic, X. Hou, S. J. Weroha, P. D. Robbins, L. J. Niedernhofer, S. Khosla, T. Tchkonia and J. L. Kirkland, Nat. Med., 2018, 24, 1246–1256 CrossRef CAS PubMed.
- S. Chikara, L. D. Nagaprashantha, J. Singhal, D. Horne, S. Awasthi and S. S. Singhal, Cancer Lett., 2018, 413, 122–134 CrossRef CAS PubMed.
- M. Daker, S. Bhuvanendran, M. Ahmad, K. Takada and A. S. Khoo, Mol. Med. Rep., 2013, 7, 731–741 CrossRef CAS PubMed.
- J. J. Hyun, H. S. Lee, B. Keum, Y. S. Seo, Y. T. Jeen, H. J. Chun, S. H. Um and C. D. Kim, Gut Liver, 2013, 7, 739–746 CrossRef CAS PubMed.
- F. Khan, K. Niaz, F. Maqbool, F. Ismail Hassan, M. Abdollahi, K. C. Nagulapalli Venkata, S. M. Nabavi and A. Bishayee, Nutrients, 2016, 8, 529 CrossRef PubMed.
- P. Ríos-Marco, C. Marco, X. Gálvez, J. M. Jiménez-López and M. P. Carrasco, Biochim. Biophys. Acta, Biomembr., 2017, 1859, 1657–1667 CrossRef PubMed.
- P. A. Jaffrès, C. Gajate, A. M. Bouchet, H. Couthon-Gourvès, A. Chantôme, M. Potier-Cartereau, P. Besson, P. Bougnoux, F. Mollinedo and C. Vandier, Pharmacol. Ther., 2016, 165, 114–131 CrossRef.
- F. Widmer, L. C. Wright, D. Obando, R. Handke, R. Ganendren, D. H. Ellis and T. C. Sorrell, Antimicrob. Agents Chemother., 2006, 50, 414–421 CrossRef CAS PubMed.
- C. Gajate, M. Matos-da-Silva, El-H. Dakir, R. I. Fonteriz, J. Alvarez and F. Mollinedo, Oncogene, 2012, 31, 2627–2639 CrossRef CAS.
- E. J. North, D. A. Osborne, P. K. Bridson, D. L. Baker and A. L. Parrill, Bioorg. Med. Chem., 2009, 17, 3433–3442 CrossRef CAS PubMed.
- A. R. D. Delbridge, S. Grabow, A. Strasser and D. L. Vaux, Nat. Rev. Cancer, 2016, 16, 99–109 CrossRef CAS PubMed.
- J. Wu, T. Liu, Z. Rios, Q. Mei, X. Lin and S. Cao, Trends Pharmacol. Sci., 2017, 38, 226–256 CrossRef CAS PubMed.
- R. Gomez-Pastor, E. T. Burchfiel and D. J. Thiele, Nat. Rev. Mol. Cell Biol., 2018, 19, 4–19 CrossRef CAS PubMed.
- V. Baylot, C. Andrieu, M. Katsogiannou, D. Taieb, S. Garcia, S. Giusiano, J. Acunzo, J. Iovanna, M. Gleave, C. Garrido and P. Rocchi, Cell Death Dis., 2011, 2, e221 CrossRef CAS PubMed.
- R. Kanagasabai, K. Karthikeyan, K. Vedam, W. Qien, Q. Zhu and G. Ilangovan, Mol. Cancer Res., 2010, 8, 1399–1412 CrossRef CAS PubMed.
- C. H. Jung, I. Cho, J. Ahn, T. I. Jeon and T. Y. Ha, Phytother. Res., 2013, 27, 139–143 CrossRef CAS PubMed.
- P. Ríos-Marco, J. M. Jiménez-López, C. Marco, J. L. Segovia and M. P. Carrasco, J. Pharmacol. Exp. Ther., 2011, 336, 866–873 CrossRef PubMed.
- M. H. Pan, Y. C. Tung, G. Yang, S. Li and C. T. Ho, Food Funct., 2016, 7, 4481–4491 RSC.
- C. E. Geisler and B. J. Renquist, J. Endocrinol., 2017, 234, R1–R21 CAS.
- B. Wang and P. Tontonoz, Nat. Rev. Endocrinol., 2018, 14, 452–463 CrossRef CAS PubMed.
- B. Gross, M. Pawlak, P. Lefebvre and B. Staels, Nat. Rev. Endocrinol., 2017, 13, 36–49 CrossRef CAS PubMed.
- L. Luo and M. Liu, J. Endocrinol., 2016, 231, R77–R99 CAS.
- C. Y. Lin and J.-Å. Gustafsson, Nat. Rev. Cancer, 2015, 15, 216–224 CrossRef CAS PubMed.
- O. Pleniceanu, R. Shukrun, D. Omer, E. Vax, I. Kanter, K. Dziedzic, N. Pode-Shakked, M. Mark-Daniei, S. Pri-Chen, Y. Gnatek, H. Alfandary, N. Varda-Bloom, D. D. Bar-Lev, N. Bollag, R. Shtainfeld, L. Armon, A. Urbach, T. Kalisky, A. Nagler, O. Harari-Steinberg, J. L. Arbiser and B. Dekel, EMBO Mol. Med., 2017, 9, 508–530 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9cc04084f |
| ‡ ZZ, BL and XL contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |